Next-generation discovery proteomics in preclinical and translational research is an exciting application of this proteomic approach. By leveraging advanced technologies, the ability to identify and quantify thousands of proteins in an unbiased manner can have a significant impact on the overall development of novel therapies.
In this blog post, we explore not only the value of using liquid chromatography-tandem mass spectrometry (LC-MS/MS) in discovery proteomics, but also the added value of incorporating ion mobility spectrometry (IMS) and data-independent acquisition (DIA) strategies.
The Power of LC-MS/MS and Proteomics in Preclinical Development
Shotgun proteomics is the practice of detecting proteins in complicated mixtures by employing bottom-up proteomics methods. In this approach, proteins are digested into peptides through enzymatic cleavage and then separated in LC prior to MS analysis.
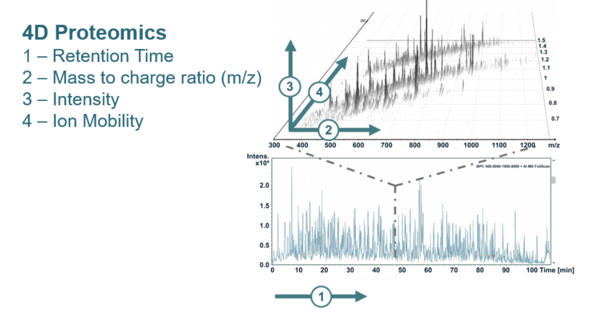
There are typically three dimensions in a bottom-up proteomics by LC-MS/MS. Retention time (RT) is one dimension, and this translates to the amount of time it takes a solute to move through the column after it has been injected. The second dimension is the mass to charge ratio of the peptide, while the third is the intensity of the spectrum peaks.
LC-MS/MS is making significant contributions to both preclinical and clinical research (see here and here for examples). Specifically, it is being used in large-scale discovery proteomics for identifying and quantifying proteins—including post-translational modifications—that are differentially expressed across biological samples. For instance, it is being used for:
Global proteomics profiling of cells, biological fluids or tissues (+/- treatment)
Biomarker discovery and validation
Drug mechanism of action and toxicity studies
Disease mechanism studies
Target identification and validation
This large-scale approach generates highly robust and unbiased datasets that capture differences across all proteins simultaneously, as opposed to conventional techniques that focus on the detection and quantitative measurement of single proteins by using ligand-binding assays. It is important to note that effective use of this technique requires a multi-disciplinary team with expertise in analytical chemistry, biochemistry, and computational biology.
What Ion Mobility Spectrometry Adds to LC-MS/MS
Ion mobility spectrometry (IMS) is an analytical technique used to separate and identify ionized molecules in the gas phase based on their mobility in a carrier buffer gas. IMS integrates seamlessly with a typical LC-MS/MS process for IM-MS, effectively adding a fourth dimension of separation.
The figure below illustrates the four dimensions of separation when using IM-MS.
This additional dimension significantly enhances scanning speed, detection sensitivity, and proteomic analysis performance in terms of identification depth, detection cycle, and quantitative accuracy. This is a great benefit when analyzing complex samples and identifying low-abundance proteins.
Moreover, IM-MS allows for DIA, which means that the MS divides precursor ions into pre-defined isolation windows and then fragments them, followed by analysis of the fragments in each window by MS. Combining IMS and DIA strategies into LC-MS/MS is becoming increasingly common since doing so further improves sensitivity, detection depth, accuracy, and data reliability.
Reduction in sample complexity and background noise since co-eluted species are separated.
Peak capacity is maximized by separating structural isomers, isobaric species, and species with differing charge states. Moreover, IM-MS can analyze both complex mixtures and single molecules since it combines broad analysis with precise targeting.
Seamless integration with other techniques (e.g., GC, LC) without disrupting the advantages of other processes in the workflow, such as chromatography and MS.
Information on an analyte’s structure via high precision collision cross section (CCS) values. The CCS measure enhances confidence when it comes to identifying molecules, reduces interference, and improves signal-to-noise ratios.
Developing insights into the structure and shape of molecules by comparing theoretical to experimental CCS values. This is important when it comes to developing insights into potential mechanisms of molecules of interest.
The Value of a DIA Strategy
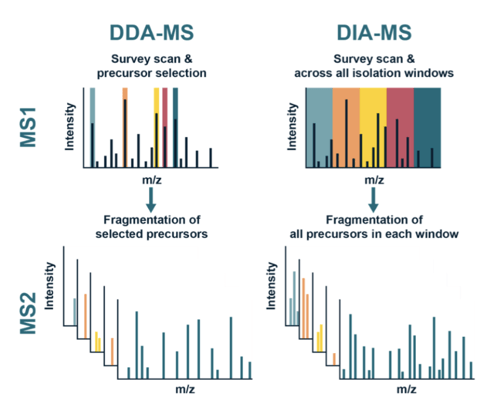
DIA (Data Independent Acquisition) and DDA (Data Dependent Acquisition) are two common modes of MS data acquisition.
In DDA mode, the instrument selects which peptides to analyze based on the abundance of their precursor ions. The instrument will typically only analyze the most abundant ions in the sample and will skip over lower abundance ions. Not only DDA can miss low-abundance ions and over present high-abundance species, it also suffers from batch effect, where the results of experiments performed at different times or on different samples can be influenced by different levels of the most abundant species in each sample.
DIA, on the other hand, is a method of acquiring mass spectrometry data in which the instrument operates the same way for all peptides in the sample, regardless of their abundance. This results in the acquisition of mass spectra for all peptides, including low-abundance species, in the sample. Unbiased DIA strategies have gained popularity in the field of quantitative proteomics, because of its ability to provide comprehensive peptide coverage.
A graphical depiction of the key differences between DDA and DIA strategies is shown below.
Benefits of an IMS–Enhanced LC-MS/MS and DIA Strategy
Integrating the use of IMS and a DIA strategy in LC-MS/MS greatly improves sensitivity, detection depth, accuracy, and data reliability, and this approach is gaining popularity. As noted earlier, incorporating these elements into an LC-MS/MS workflow provides an additional dimension of separation and allows for deeper analysis of samples, with one study indicating that DIA identified between 2 and 3.3 times more proteins as compared to DDA, and another study reporting that more than 10,000 proteins were identified from a single cell type in 2 hours or less without prior MS measurements for spectral library generation.
Conclusion
Next generation IM-MS-based-proteomics in combination with a DIA strategy is enabling powerful proteomic analyses in preclinical and translational research for understanding the biological function of proteins, biomarker identification and validation, global proteomics profiling, and target identification and validation, among other uses.
Han, Y., (2023) Next-Generation Discovery Proteomics: The Power of Ion Mobility Mass Spectrometry with Data-Independent Acquisition - Crown Bioscience. https://blog.crownbio.com/next-generation-discovery-proteomics
Spatial biology is transforming our understanding of tissue microenvironments, providing unprecedented insights into gene expression and protein inter…
Gene editing has become a cornerstone of modern molecular biology, with applications ranging from basic research to clinical therapies. While traditio…
The speed at which we can translate scientific discoveries in oncology research into tangible treatments is crucial. The future of cancer treatment li…