Explore the ‘undruggable’ oncology target RAS, inhibitory techniques used on downstream pathways, and the preclinical models available to test up-and-coming RAS targeted agents.
One-third of cancers diagnosed each year are driven by mutations in RAS family genes, including 95% of pancreatic cancers and 45% of colon cancers. This, in theory, presents an attractive family of targets for treating many cancer types.
The RAS family is represented by three members:
KRAS, the most frequently mutated (85% of all RAS-driven cancers).
Followed by NRAS (12%).
And HRAS (3%).
All three RAS proteins share over 80 percent of their amino acid sequence, with mutations occurring predominantly in three genes, in codons 12, 13, and 61.
RAS Proteins and Cell Proliferation
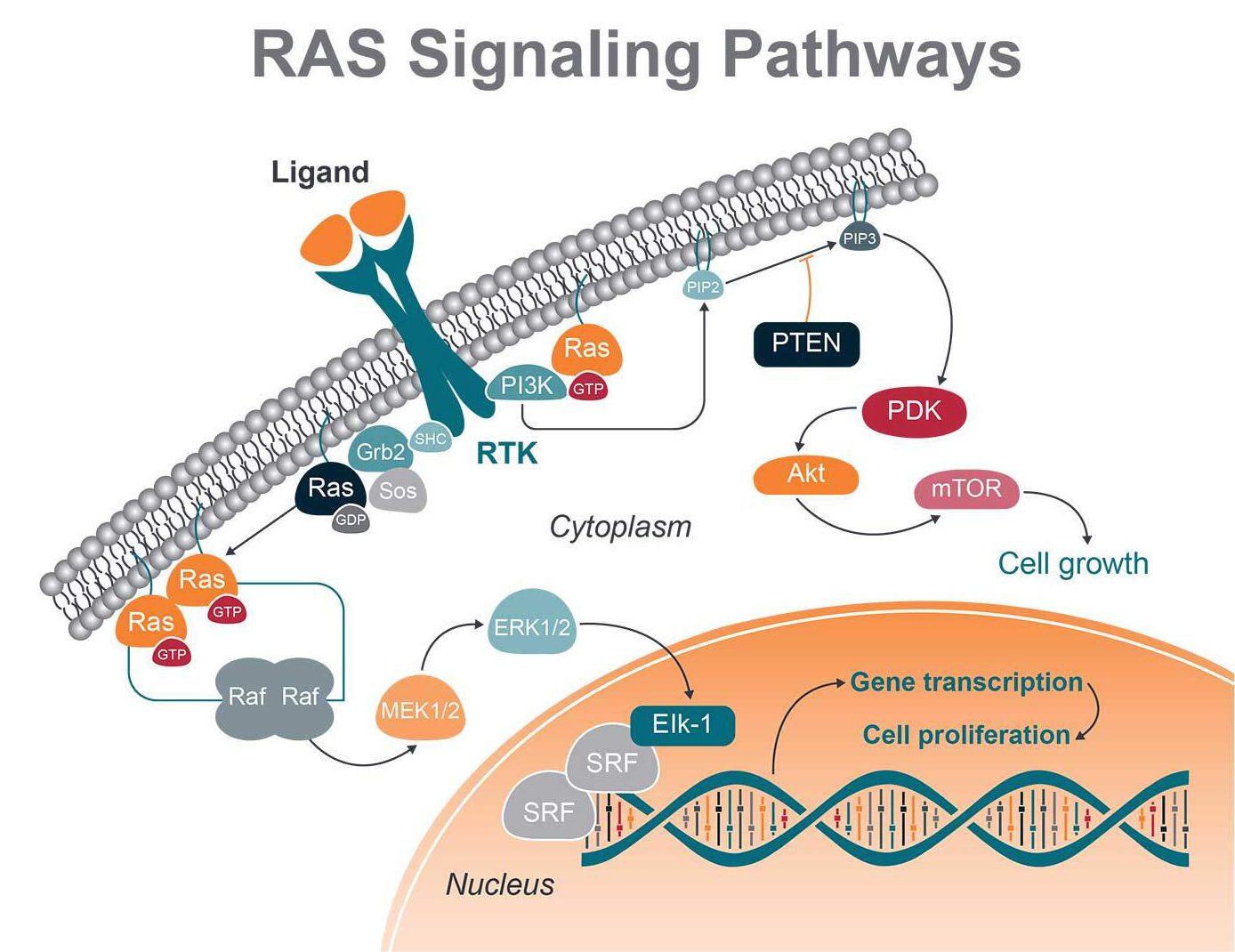
RAS proteins are membrane-associated G proteins, meaning they can bind GTP and GDP. When RAS is bound to GTP, the protein is switched to an active state, inducing cell growth, proliferation, and migration. GDP binding switches the protein to an inactive state.
When RAS family members are mutated, they are permanently bound to GTP, driving cell proliferation continuously.
RAS: An Undruggable Oncology Target
One solution to stop oncogenic RAS would be to block its activation. However, attempts to develop drugs that target mutant RAS proteins have been unsuccessful.
This is due to the relative cellular abundance of GTP and the binding affinity of RAS for GTP being extremely high. There’s also an apparent lack of suitable surfaces in critical regions of RAS proteins necessary for small molecules to bind, making tumors bearing these mutations among the most difficult to treat. RAS-related treatment strategies instead use an indirect approach, targeting pathways downstream of RAS.
Downstream RAS Pathway Drug Targets
RAS-RAF-MEK-ERK
One important signaling route is the RAS-RAF-MEK-ERK (MAPK) cascade, initiated by ligand binding to receptor tyrosine kinases (RTK). Once RAF activation takes place, RAF phosphorylates and activates mitogen-activated protein kinases 1 and 2 (MEK1 and MEK2). These proteins can then both activate the downstream extracellular signal-regulated kinases 1 and 2 (ERK1 and ERK2).
ERK 1 and 2 regulate transcription factors in the nucleus (i.e., c-Myc, c-Fos, and CREB) via phosphorylation, acting on gene expression involved in cellular growth, cell differentiation, and division.
The recent approval of KRAS G12C inhibitors, sotorasib (AMG510) in 2021 and adagrasib (MRTX849) in 2022, for the treatment of NSCLC, has revived interest in developing KRAS inhibitors. Beyond KRAS G12C mutations, specific inhibitors targeting other KRAS are under development. For example, MRTX1133, has been shown to bind KRAS G12D in a non-covalent binding form (KD = ∼0.2 pM) with a binding selectivity which is 700-fold higher compared to KRAS WT. TKR15 could bind to KRAS G12V protein and significantly inhibit the proliferation of A549 cells with IC50 of 0.21 µM. A series of small molecules that irreversibly bound to KRASG12S and suppressed its oncogenic signaling without affecting its wild-type counterpart in KMS20 cells have been described. Recently, it was shown that BI-2865, as a pan-KRAS inhibitor, can prevent the activation of wild-type KRAS and target a broad range of KRAS mutants, including G12A/C/D/F/V/S, G13C/D, V14I, L19F, Q22K, D33E, Q61H, K117N and A146V/T. Additionally, BRAF inhibitors, vemurafenib and dabrafenib, and MEK inhibitors, trametinib and cobimetinib, have been approved for BRAF V600E/K metastatic melanomas.
PI3K-Akt
RAS also interacts with and stimulates the PI3K-Akt pathway. Once PI3K is activated by RAS, AKT is in turn phosphorylated and activated, with a number of downstream effects. These include activation of mTOR, which regulates the protein synthesis necessary for cell growth, proliferation, and angiogenesis.
The PI3K pathway has also been selected as a target for cancer therapy due to the discovery of overactivation of PI3K in a variety of cancers and its significance for the proliferation and survival of cancer cells. However, in the course of treatment, problems such as abnormal activation of feedback, compensation activation, drug resistance, and toxicity of PI3K pathway inhibitors are found.
Current Inhibitory Routes: BRAF and MEK
Targeting MAPK signaling using BRAF inhibitors (e.g., vemurafenib, dabrafenib) and/or MEK inhibitors (e.g., trametinib, cobimetinib) has proven to be an effective treatment for a variety of different cancers. Resistance remains a major challenge, however, 30% of tumors don’t respond to the inhibitors, and progression is often observed within the 70% that do respond.
Preclinical Models for RAS Targeted Therapies
A variety of preclinical models are available for assessing agents targeting RAS and downstream pathways, including gene-engineered human cell-line derived xenografts (CDX), genetically engineered mouse models (GEMM), and patient-derived xenografts (PDX).
Genetically Engineered Cell Line Derived Mouse Models
In the preclinical stage, human cell lines can be genetically modified with gene mutations, and human CDX can be established using them.
Primary or secondary KRAS mutant tumor cell lines can be generated for testing therapies targeting the RAS oncogene family. These CDX models have been successfully used for evaluating the efficacy of KRAS, BRAF, MEK, and mTOR inhibitors both as single agents and in combinations.
For instance, the HCT116 cell line, which harbors a KRAS G13D mutation, and the A549 cell line with a KRAS G12S mutation, offer valuable insights into the dynamics of KRAS-driven carcinogenesis and the potential efficacy of targeted therapies. Similarly, the MiaPaCa-2 (KRAS G12C) and SW48 (KRAS G12V) cell lines serve as critical models for assessing the pharmacodynamics and therapeutic windows of novel KRAS inhibitors.
These twostudies highlight the complexities and therapeutic opportunities within the spectrum of KRAS mutations, demonstrating the critical role of model systems like CDX in driving forward clinical applications.
Learn about how we established KRAS G12C Inhibitor-resistant tumor models in our AACR 2024 poster presentation.
Genetically Engineered Mouse Models
Preclinical GEMM recapitulates disease characteristics, where genes that are strongly associated with tumor progression and development are deleted, overexpressed, or mutated. This results in spontaneous tumor formation.
There’s a large collection of GEMM available covering many common cancer indications, such as lung, prostate, breast, colon, and pancreatic cancers.
KRAS mutant GEMM are of particular interest for testing therapies targeting the RAS oncogene family. Indeed, conditional expression of KRAS mutants in the lung, pancreas, and gastrointestinal tract induces preneoplastic epithelial hyperplasias, adenomas, pancreatic intraepithelial neoplasia, and adenocarcinomas. These models have been successfully used to demonstrate the efficacy of BRAF, MEK and mTOR inhibitors both as single agents and in combinations.
Patient-Derived Xenografts
PDX are also valuable and translational preclinical models to evaluate RAS-targeting therapies. PDX are animal models derived directly from patient tissue samples, which maintain genotypic and phenotypic fidelity to the patient from whom they were derived. This provides more predictive models than traditional xenografts.
Because of their temporal proximity to the patient, and having never been subjected to the selection pressures of cell culture, PDX models closely recapitulate patient disease. These models show high fidelity in the histological presentation of the patient’s cancer, but also in response to chemotherapy, radiotherapy, and targeted therapies.
RAS proteins are essential components of the signaling networks controlling cell growth, proliferation, and migration. RAS mutations are frequently found in human tumors and are largely recalcitrant to targeted therapies.
GEMM and PDX RAS mutant models recapitulating critical aspects of human disease provide important models for oncology research and are widely used for preclinical validation of targets and therapies.
Cite this Article
Hua, A., (2024) RAS: Targeting the Impossible - Crown Bioscience. https://blog.crownbio.com/targeting-ras-oncology
Overcoming drug resistance presents a significant challenge in cancer treatment for both solid and liquid tumors. A comprehensive collection of resist…
In the dynamic field of oncology research, KRAS has long been a formidable opponent. Originally deemed undruggable, recent breakthroughs have now posi…
The intricate relationship between the microbiome and cancer has emerged as a transformative area of study in oncology. While advances in immuno-oncol…