Each year, around 140,000 people worldwide (15,000 in the United States alone) are diagnosed with epidermal growth factor receptor (EGFR)-positive non-small cell lung cancer (NSCLC). EGFR-positive cancers have genetic changes that lead to an overactive EGFR protein, which fuels the growth of cancer cells.
Different mutations result in different treatment paths, and this post looks at the therapeutics available for classical, acquired resistance, and non-canonical mutations.
EGFR Mutations are NSCLC Oncogenic Drivers and therefore, Drug Targets
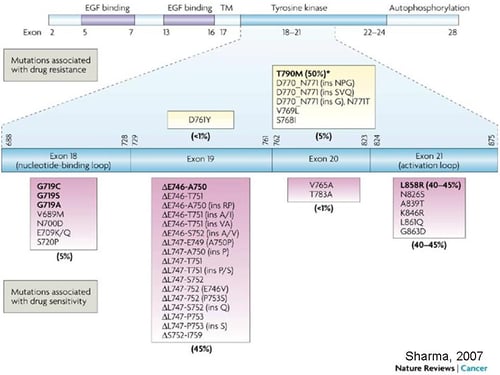
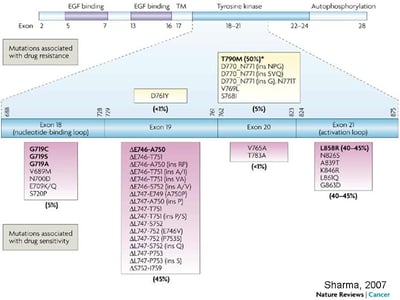
Activating EGFR mutations are found as oncogenic drivers in a significant proportion of NSCLC cases (∼15% of all NSCLC). The range of EGFR genetic alterations include:
the classic exon 19 in-frame deletion (45%)
exon 21 L858R (40%)
the exon 20 T790M “gatekeeper” mutation
as well as the non-canonical exon 18 G719X (3%) and exon 20 insertions (5–10%)1.
All of these EGFR genetic alterations are regarded as important drug targets for NSCLC.
Tyrosine Kinase Inhibitors are Useful for Patients with Classical EGFR Activating Mutations
NSCLC patients with classic EGFR activating mutations are usually responsive to particular EGFR tyrosine kinase inhibitors (TKIs). It’s been nearly 15 years since EGFR-targeted therapies were first introduced, helping to extend survival for thousands of patients since their approval.
The first generation EGFR TKIs - gefitinib, erlotinib, and icotinib - were designed to reversibly combine with adenosine triphosphate (ATP) binding sites and therefore block EGFR-induced activation of downstream signaling, inhibiting both wild-type and mutated EGFR.
Despite this, first generation TKIs have a higher binding affinity to mutated EGFR, ensuring a potent response.
TKI Resistance Emerges to Reduce Survival Benefits
First generation TKIs have been extensively investigated in NSCLC harboring activating EGFR mutations (deletions in exon 19, and exon 21 L858R mutation).
Compared with chemotherapy as first-line treatment of advanced NSCLC harboring EGFR mutations, the first generation EGFR TKIs significantly improved response rate (RR) and progression-free survival (PFS).
However, overall survival (OS) was not improved as the EGFR T790M resistance mutation ultimately emerged in most of these patients.
More Potent Second Generation EGFR TKIs
In contrast, the second generation (afatinib or dacomitinib) or third generation EGFR TKIs (osimertinib, rociletinib, and HM61713) were designed to have more potent inhibition of EGFR, as well as to overcome EGFR T790M.
The second-generation EGFR-TKIs are irreversible inhibitors with greater affinity for the EGFR kinase domain, also inhibiting other members of the EGFR family (ErbB2, ErbB3, and ErbB4).
Third Generation EGFR TKIs Overcome the Gatekeeper T790M Mutation, But Resistance Still Occurs
Third generation TKIs are active against EGFR-activating mutations and the T790M resistance mutation, and have only limited efficacy against wild-type EGFR.
However, despite the promise of this last generation of TKIs, resistance has been described with C797X mutation identified as one primary mechanism.
Monoclonal Antibodies Also Target Classic EGFR Mutations
Cetuximab (Erbitux®), a monoclonal antibody that interacts with the extracellular domain of EGFR, also demonstrated impressive activity against NSCLC with classic EGFR mutations2, with an assumed mechanism of blocking dimerization and/or ligand binding3.
What about Treating Patients with Non-Canonical EGFR Mutations?
The efficacy of the drugs discussed so far on NSCLC harboring non-canonical EGFR mutations is a lot less clear1. Exon 20 insertions are the most prevalent of the non-canonical EGFR mutations, located close to the end of the regulatory C-helix domain within the N-lobe of the kinase (after M766). These types of mutation are generally associated with resistance to existing TKIs (gefitinib, erlotinib, and afatinib)1,4,5, and the basis of this de novo resistance is poorly understood.
Further Preclinical Models Needed to Fully Understand and Overcome Resistance Mechanisms
Today, the lack of preclinically relevant models recapitulating patient non-canonical genotypes (either patient-derived xenografts, PDX, or genetically engineered mouse models), is a major obstacle for studying exon 20 insertion responses to treatment5. Most of the currently available experimental data on exon 20 insertion mutant response to TKIs is derived from in vitro artificially created surrogate cell line models1, with a small number of unique PDX models available5.
As more mechanisms of de novo and acquired resistance across all of NSCLC are unraveled, clinically relevant and predictive preclinical models are required to fully understand them, and to evaluate new agents and combination therapies to overcome this resistance, as well identifying which patients will benefit from any given treatment regimen.
References and Further Reading on the topic can be found here:
Overcoming drug resistance presents a significant challenge in cancer treatment for both solid and liquid tumors. A comprehensive collection of resist…
In the dynamic field of oncology research, KRAS has long been a formidable opponent. Originally deemed undruggable, recent breakthroughs have now posi…
The intricate relationship between the microbiome and cancer has emerged as a transformative area of study in oncology. While advances in immuno-oncol…


 EGFR Mutations in Lung Cancer and Their Effect on Drug Response
EGFR Mutations in Lung Cancer and Their Effect on Drug Response