Discover the complex range of FGFR family aberrations in cancer and how PDX models are a powerful tool for assessing new FGFR-targeted therapeutic options.
The Multiple Pathways Involved in Cancer Development
Aberrant receptor tyrosine kinase signaling leads to hyperactivation downstream of many pathways implicated in oncogenesis. One of these is the RAS pathway; we’ve previously discussed why mutations in RAS family genes are important in cancer development and how to target this therapeutically.
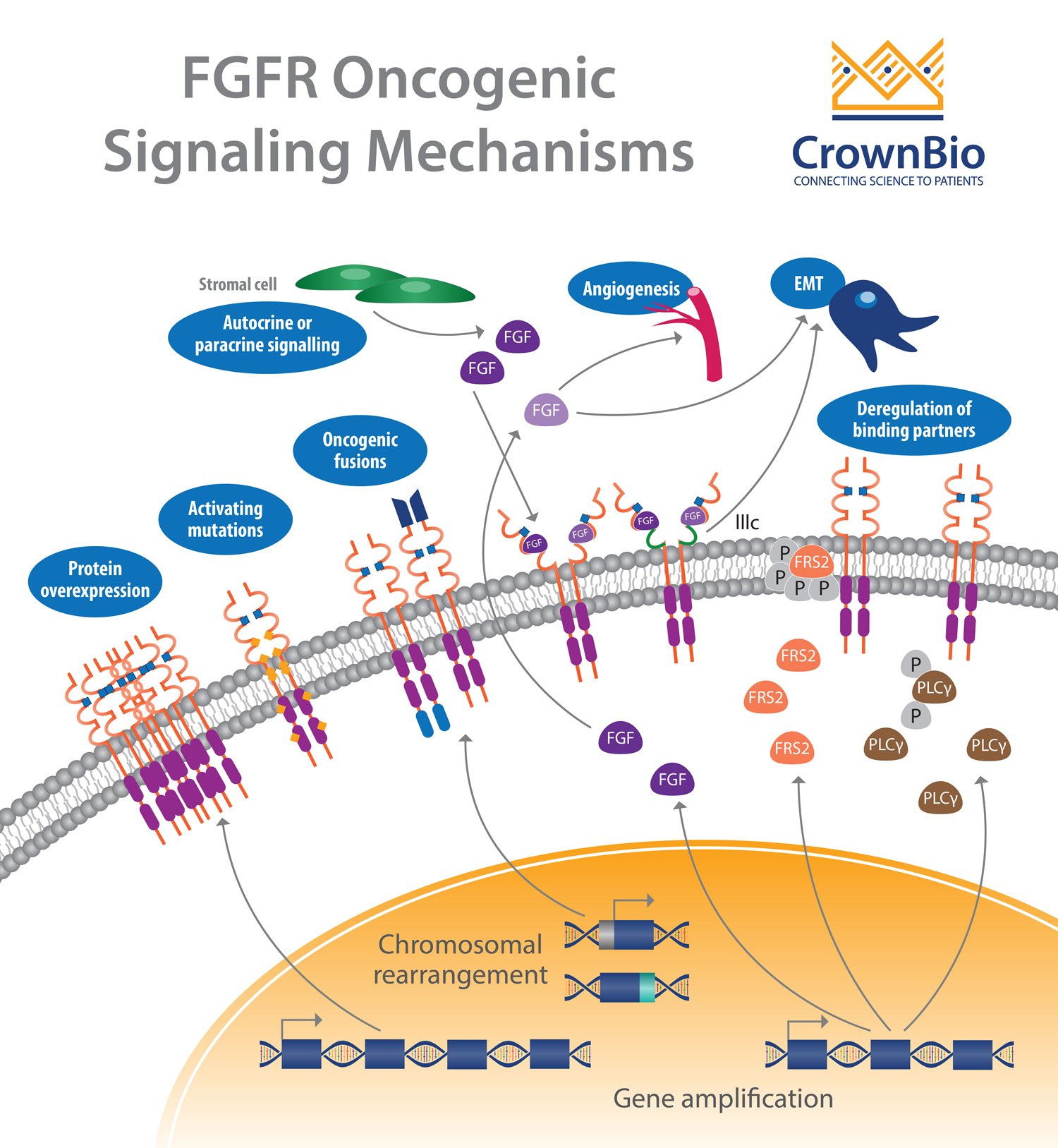
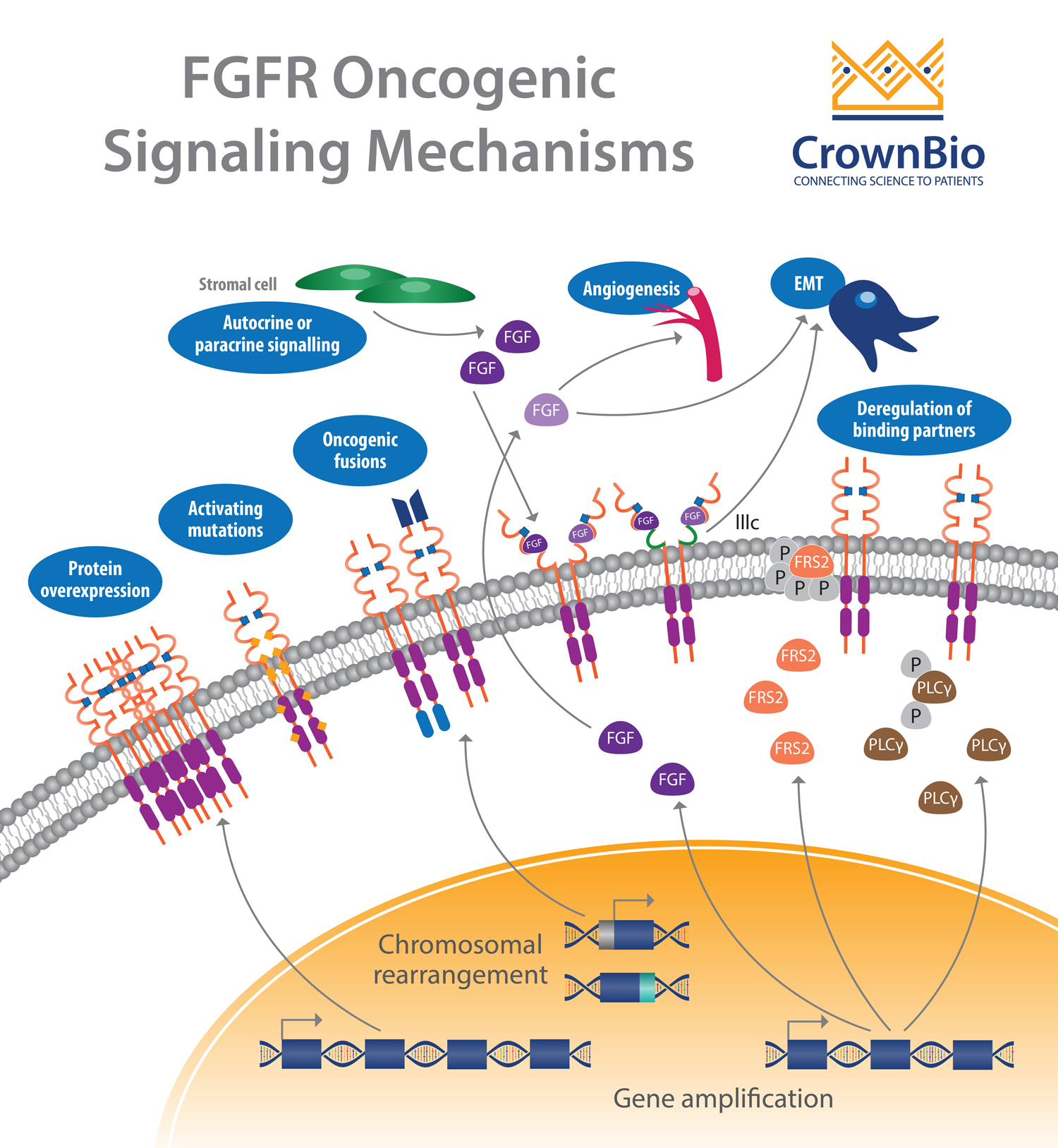
Another affected pathway centers around the fibroblast growth factor receptor (FGFR) which is frequently altered in human cancer. Deregulation of the fibroblast growth factor (FGF)/FGFR signaling axis is implicated in oncogenesis, tumor progression, and resistance to anticancer therapy across many tumor types.
FGFR activation by FGF leads to phosphorylation of specific tyrosine residues. This initiates a cascade of intracellular events activating the major survival and proliferative signaling pathways RAS-MAPK, PI3K-AKT, PLCγ, and STAT.
FGFR Aberrations in Cancer
An extensive review of the range of FGFR aberrations found across different cancer types has been performed, with over 4,850 tumors analyzed by Next-Generation Sequencing (NSG).
Overall, 7% of the tumors tested had FGFR aberrations, split proportionally as:
FGFR1 - 49%
FGFR2 - 19%
FGFR3 - 23%
FGFR4 - 7%
Gene amplifications provided the majority of the FGFR aberrations (66%), followed by mutations (26%) and rearrangements (8%).
Almost every cancer type showed some tumors with FGFR aberrations. Higher frequencies were observed in urothelial cancer (32%), breast cancer (18%), endometrial cancer (∼13%), squamous lung cancers (∼13%), and ovarian cancer (∼9%).
FGFR Amplifications
FGFR1 amplifications (8p12 locus) are found in approximately 15% of squamous non-small cell lung cancers (NSCLC) and 10% of breast cancers (predominantly in hormone receptor positive disease). They are also observed to a lesser extent in ovarian, bladder, renal cell carcinoma, and rhabdomyosarcoma.
Amplification of FGFR3 and FGFR4 is not frequently reported, with activation of these receptors often linked to somatic mutation.
FGFR Mutations
Somatic activating FGFR point mutations have been identified, resulting in multiple aberrant signaling mechanisms. Most of these mutations are frequently observed outside the kinase domain, which is in opposition to other tyrosine kinase receptors (e.g. EGFR). Mutations in FGFR2 and FGFR3 are the more frequent with rare mutations observed for FGFR1.
FGFR2 mutations, consisting mainly of missense activating mutations (S252W, P253R), are found in around 12% of endometrial cancer, with lower proportions seen in NSCLC, gastric, and urothelial cancer.
FGFR3 mutations are found in around 75% of invasive urothelial cell carcinomas, 15% of high-grade invasive urothelial cancers, and around 5% of cervical cancers.
Finally, FGFR4 mutations (K535, E550) present in the kinase domain and which induce autophosphorylation of the receptor are found in 7% of rhabdomyosarcoma.
Gene fusions are the result of translocation, interstitial deletion, or chromosomal inversion. FGFR gene fusions occur at a low incidence, with fusions involving FGFR2 and FGFR3 the most common.
The discovery of the first recurrent FGFR fusion, FGFR3-TACC3, was observed in high-grade brain tumors (i.e. glioblastoma), followed by bladder and, in some cases, lung cancer.
FGFR1 fusions although non-recurrent (i.e. ZNF198, BCR, FOP) have been observed mostly in myeloproliferative disorder stem cell leukaemia/lymphoma syndrome (SCLL/8p11 myeloproliferative syndrome) but also in in lung squamous cell carcinoma (LUSC), breast cancer, glioblastoma and low-grade brain tumors.
FGFR2 fusions (i.e. CIT, CCDC6, CCAR2, OFD1, BICC1, etc.) are found in 15% of intrahepatic cholangiocarcinoma and on rare occasions in thyroid, lung, and prostate cancers.
Deregulation of Autocrine/Paracrine Loops
In addition to genetic abnormalities which lead to ligand-independent signaling and constitutive receptor activation, ligand-dependent signaling involving FGFs and FGFRs can also be deregulated.
More recently, multiple FGFs have been described in various cancers including breast, prostate, colorectal cancers, hepatocellular carcinoma, and NSCLC.
Targeting the FGFR Family Therapeutically
In light of the many alterations in the FGF/FGFR pathways, multiple therapeutic strategies have been developed or are in development. These strategies use non-selective and selective FGFR tyrosine kinase inhibitors (TKIs), antagonist antibodies, or FGF ligand traps.
Non-selective FGFR TKIs (e.g. dovitinib, lenvatinib, lucitanib, nintedanib) bind to the ATP-binding domain of the catalytic site of the receptor, but can also target other RTKs (i.e. VEGFR and PDGFR).
In order to increase specificity, and reduce the toxicity associated with multitargeting TKIs, selective FGFR inhibitors have been developed including compounds like AZD4547, NVP-BGJ398, JNJ-42756493, LY2874455, TAS120, and Debio-1347.
Several monoclonal antibodies targeting either FGF/FGFR are in preclinical or early phase development. These antibodies block FGFR signaling by interfering with ligand binding or receptor dimerization and include antiFGFR3 (MGFR1877S), anti-FGFR2-IIIb (GP369, FPA144), and anti-FGFR2 (BAY1187982).
The final strategy is to prevent FGF ligand binding to FGFR. The soluble fusion protein FP-1039 (GSK3052230) consists of the extracellular domain of FGFR1c fused to the Fc region of IgG1 preventing FGF1, FGF2, and FGF4 binding. A Phase IB trial has been completed evaluating FP-1039 in association with chemotherapy in patients with lung cancer (NCT01868022).
Evaluating FGFR-Targeted Therapies Preclinically with PDX Models
While multiple strategies are being developed, clinical response has so far been limited. In order to better evaluate the different FGFR-targeted therapeutic strategies, improved preclinical models are needed. More predictive models which fully capture the complex interplay and mechanism of the different FGFR clinical aberrations are required.
Patient-derived xenografts (PDX), which are highly clinically-relevant preclinical models recapitulating human disease, provide a powerful approach. Models are available featuring clinically relevant FGFR aberrations (such as FGFR3 fusions, including FGFR3-TACC3). Databases collating PDX NGS data also allow the search and selection of models featuring rare FGFR fusions or mutation which may be hard to find in traditional CDX models.
Numerous publications have demonstrated the use of PDX to investigate FGFR targeted therapies in different indications, such as cholangiocarcinoma, endometrial, NSCLC, gastric and HCC cancer.
Mouse Clinical Trials
Large panels of PDX models can also be used in Mouse Clinical Trials, which mimic a patient Phase II trial. Each PDX subject reflects the pathology of its original patient, behaving as a patient avatar, and the cohort of patient avatars represents a diversity of the human patient population. This can allow testing of an agent across a population of FGFR mutated/fused disease, and to assess how different aberrations affect response.
As well as studying response to a drug across a population, another main use of PDX Mouse Clinical Trials is to identify predictive biomarkers. This has already been accomplished for dovitinib in lung squamous cell carcinoma (LSCC), showing that overexpression of gene sets associated with FGFR pathway activation could be a potential biomarker for FGFR inhibition in LSCC patients.
Summary
The FGF/FGFR signaling pathway is complex, with multiple alterations and heterogenous behaviors across a variety of cancer types. Currently, there are multiple FGFR inhibitors in development, which so far have had limited clinical response.
More clinically-relevant and predictive preclinical models, such as PDX, are required for preclinical testing. This will allow us to have a deeper understanding of the mechanisms at play, and to improve evaluation of new agents and combination therapies to overcome the currently limited efficacy. New models can also help to identify further predictive biomarkers.
Cite this Article
Bourré, L., (2019) Targeting the FGF/FGFR Signaling Axis with PDX Models - Crown Bioscience. https://blog.crownbio.com/pdx-models-targeting-fgf-fgfr
Overcoming drug resistance presents a significant challenge in cancer treatment for both solid and liquid tumors. A comprehensive collection of resist…
The Resurgence of KRAS as a Therapeutic Target In the dynamic field of oncology research, KRAS has long been a formidable opponent. Originally deemed …
The intricate relationship between the microbiome and cancer has emerged as a transformative area of study in oncology. While advances in immuno-oncol…