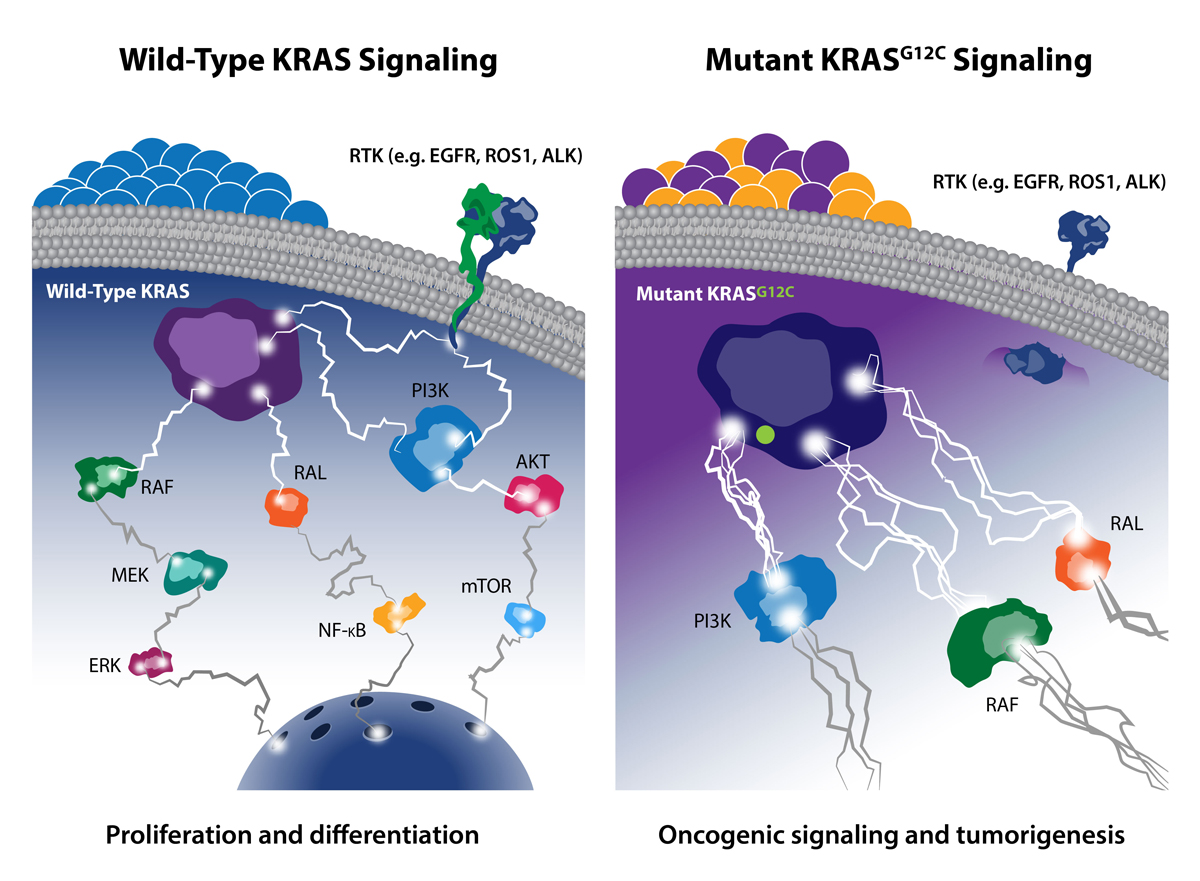
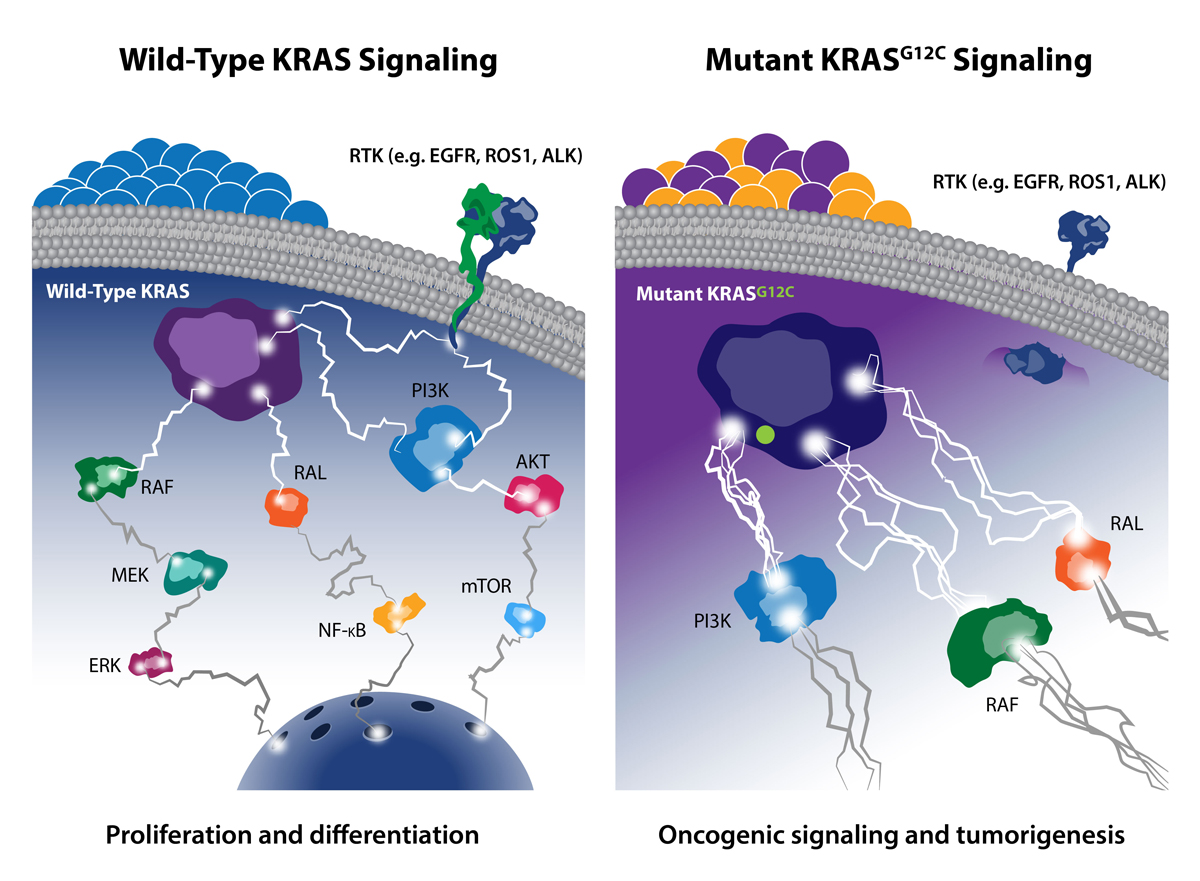
After decades of attempting to target KRAS, scientists finally succeeded in 2013 by developing a KRAS inhibitor that specifically targets the KRAS G12C mutation found in 14% of lung cancers, 3% of colorectal cancers, and 1–3% of other solid tumors.
As a previous post discussed, one of the most common KRAS gene mutations occurs at codon 12, which affects the intrinsic GTPase activity of the protein. Mutation of glycine 12 (G12) results in RAS activation by interfering with GAP binding and GAP-stimulated GTP hydrolysis. G12C is a single point mutation, substituting glycine 12 for cysteine, and is highly dominant in lung cancer, occurring in about 14% of lung adenocarcinomas. Overall, KRAS G12C accounts for over ~12% of all KRAS G12 mutations, so multiple drug developers have actively targeted it.
While researchers have long tried to use small molecules to target KRAS G12C, these were typically nonselective (they bound both the mutant and wild-type forms). However, newer small molecules have been developed to form covalent bonds with the mutant cysteine and have incredible selectivity for the mutant KRAS protein over the wild-type form. Such covalent inhibitors are thought to lock the KRAS G12C in the inactive state, therefore blocking activated oncogenic signaling. Researchers are also targeting an adjacent histidine 95 (H95) residue, which may further enhance the drug-protein interaction.
AMG 510 [Amgen] was the first KRAS G12C inhibitor to reach clinical development. Early results indicate that AMG 510 has antitumor activity in both non-small-cell lung cancer (NSCLC) and colorectal cancer. For lung cancer, the objective response rate was 48% for 23 evaluable patients who had completed the first six-week CT scan or had early progressive disease. These data resulted in the Food and Drug Administration granting fast track designation to AMG 510 for KRAS G12C-positive NSCLC.
MRTX849 [Mirati Therapeutics] is the second inhibitor to reach clinical development. MRTX849 demonstrated pronounced tumor regression in 17 of 26 (65%) KRAS G12C positive cell line and patient-derived xenograft (PDX) models from a variety of tumor types. In humans, three out of six lung cancer patients (50%) and one out of four colorectal cancer patients (25%) experienced partial tumor shrinkage, while two people with appendix cancer did not. However, those patients whose tumors did not shrink were found to have stable disease.
Despite these amazing results, there remains an unmet need for novel agents that molecularly target KRAS-mutant alleles beyond G12C—such as G12D and G12V. Since many KRAS-driven cancers rely on upstream signaling from proteins, such as SHP2 and SOS1, researchers are now targeting these factors as a new therapeutic framework.
SHP2 is a protein-tyrosine phosphatase that mediates cellular signaling through the RAS/MAP kinase pathway by activating SOS1-regulated RAS-GTP loading. Several companies are exploring SHP2 inhibitors, with the most advanced—RMC-4630 and TN0155—currently in Phase 1 trials. Published data show particular sensitivity to SHP2 inhibitors in KRAS G12C mutant tumors. Further, MRTX849 (discussed earlier) is being tested in combination with Novartis’ TNO155 SHP2 inhibitor in patients with advanced solid tumors with KRAS G12C mutations. This strategy is based on nonclinical data that showed that such a combination resulted in a significant increase in antitumor activity versus the individual agents alone.
SOS1 is activated by RAS through the binding of RAS(ON) to an allosteric site on the SOS1 protein, which results in a positive feedback loop between SOS1 and RAS to increase RAS signaling. The activation of RAS by SOS1 is “processive”: once a single molecule of SOS1 is activated, it can then sequentially activate many RAS molecules. As a result, the amplification of RAS signals by SOS1 is potentially large.
Preliminary data from Boehringer Ingelheim suggest that combined SOS1/MEK inhibition (using a SOS1:pan-KRAS inhibitor) shows great cooperativeness in multiple G12 and G13 KRAS-mutated PDX models. Further, Boehringer Ingelheim recently announced that it will begin testing its SOS1:pan-KRAS inhibitor in combination with MRTX849 for patients with lung and colorectal cancers with the KRAS G12C mutation. In preclinical studies, combining a KRAS G12C inhibitor with a SOS1:pan-KRAS inhibitor resulted in enhanced antitumor activity based on the complementary mechanisms of these agents.
Conclusion
Activating mutations in RAS are the most common driver mutations in cancer. Therefore, it is not surprising that this family of genes is an attractive target for treating many different cancer types. Incredible progress has been made in recent years by targeting specific mutant forms of KRAS, especially the G12C mutant. Additional advancements are being made through the pharmacological manipulation of upstream KRAS-mutant targets such as SHP2 and SOS1, which may lead to optimized therapeutic combinations for KRAS-mutated cancers.
Cite this Article
Bourré, L., (2020) SHP2 and SOS1 Targets: Additional Players in the KRAS Inhibitors Race? - Crown Bioscience. https://blog.crownbio.com/shp2-and-sos1-targets
Overcoming drug resistance presents a significant challenge in cancer treatment for both solid and liquid tumors. A comprehensive collection of resist…
In the dynamic field of oncology research, KRAS has long been a formidable opponent. Originally deemed undruggable, recent breakthroughs have now posi…
The intricate relationship between the microbiome and cancer has emerged as a transformative area of study in oncology. While advances in immuno-oncol…