Advances in genomic sequencing technologies have greatly aided our understanding of the microbiome and its impact on the host. In this blog post, we explore some of the common sequencing approaches used to study the complexities of the microbiome at the genome and transcriptome levels.
The Microbiome
The microbiome regulates a variety of important physiological processes, from metabolite production to immune system function. Many important studies have now associated a disrupted microbiome with diseases affecting a variety of organ systems.
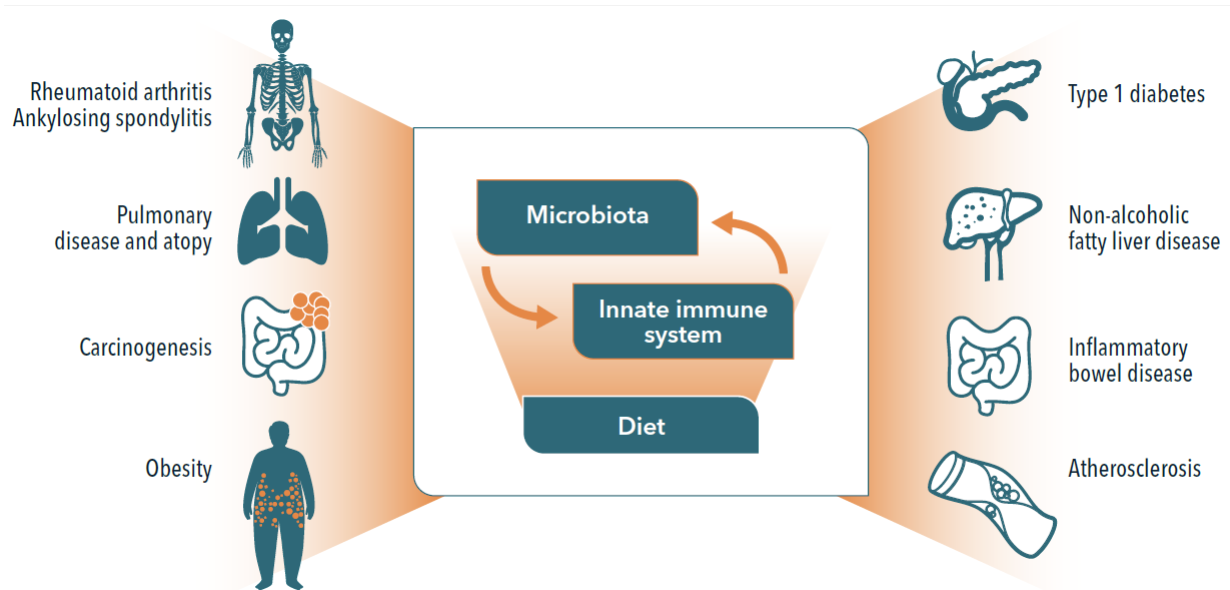
Figure 1. The link between microbiome dysbiosis and diseases affecting different organ systems
Until recently, the study of the microbiome relied heavily on culture-dependent methods, which naturally limited the analyses to microorganisms that are culturable, although many microorganisms are known to be “unculturable.” The availability of novel sequencing technologies has more recently empowered researchers to identify, classify, and quantify known and novel microbes without relying on culturing.
The two main approaches used for genomic profiling of the microbiome are 16S ribosomal RNA (rRNA) amplicon sequencing (targeted) and shotgun metagenomics (whole genome).
16S sequencing
Targeted sequencing of the 16S rRNA gene is a well-established method. This gene encodes a key component of the transcriptional machinery in bacteria and archaea and contains both highly conserved genetic sequences and hypervariable regions that exhibit diversity among species. These features make 16S an ideal target for microbiome profiling, as not only do they permit universal amplification of genomic material but also microorganism discrimination in complex samples. As a cost-effective microbiome analysis method, it is especially suitable for simple samples or samples with some pre-knowledge of content.
16S workflow and bioinformatic analysis
A basic 16S rRNA sequencing workflow would be the following:
- Sample collection
- DNA extraction
- PCR-based amplification of selected variable regions
- Sequencing
- Data analysis
Popular sequencing platforms include those that generate short-read data of approximately 150–500bp (e.g., Illumina’s MiSeq, NextSeq, and NovaSeq), and in the recent years, long-read sequencing platforms (discussed later).
Full-length 16S sequencing with third-generation sequencing (TGS) technology
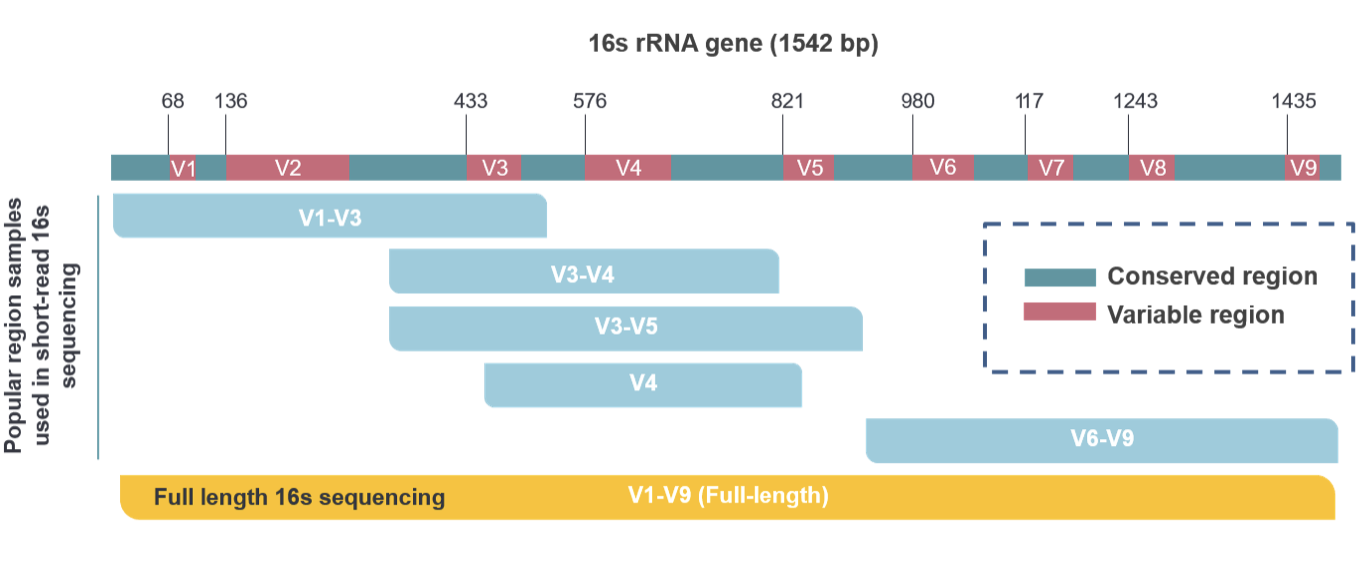
Figure 2. Commonly used DNA regions in short-read and long-read 16S rRNA sequencing
The primary benefit of TGS technology (PacBio RS II/Sequel and Oxford Nanopore MinION) over second-generation sequencing platforms is that they generate long-read data, which easily covers the relatively small 1.5 kb 16S rRNA gene and all hypervariable regions. Crown Bioscience uses the PacBio RSII/Sequel system for Hi-fi read sequencing, during which each genetic locus is circularized and repeatedly read through until a consensus sequence is reached. This is called circular consensus sequencing (CCS) mode, and it guarantees the yielded reads with a high accuracy of 99.9%.
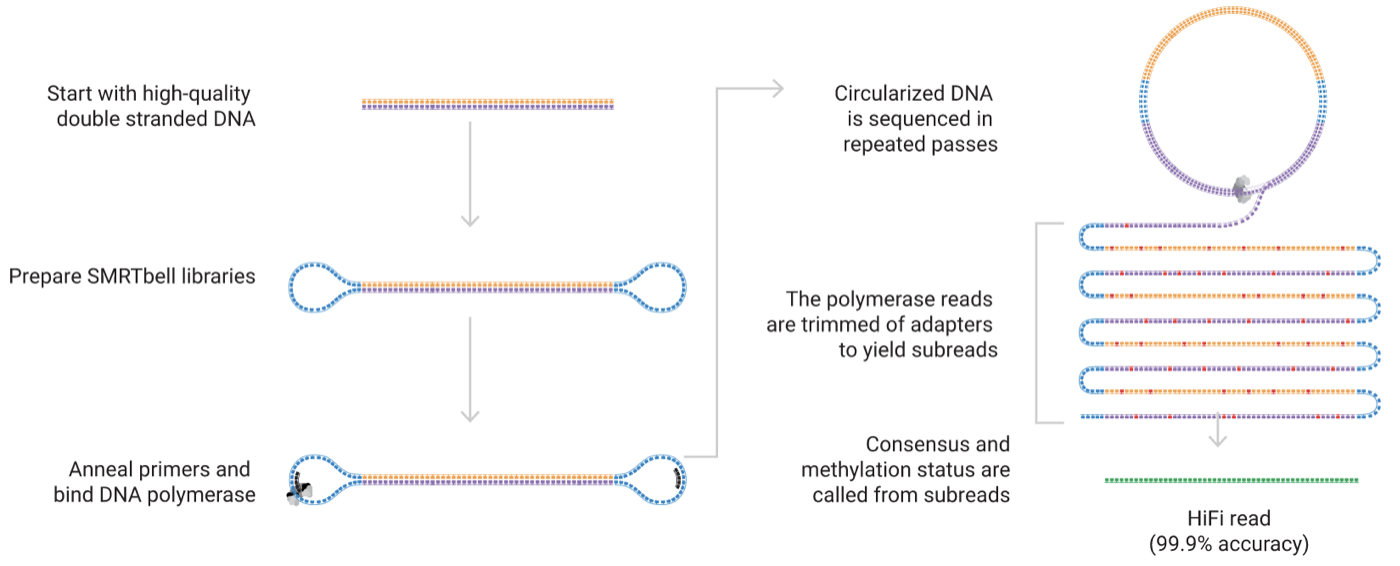
Figure 3. HiFi sequencing by circular consensus sequencing mode (Source: Pacbio)
TGS platforms overcome the limitations of traditional 16S rRNA analysis because, they only read partial regions on the 16S rRNA gene, short read 16S sequencing is not as sensitive and limited in species identification. The advantages of long-read sequencing for obtaining better taxonomic resolution have been established in published studies. For instance, multiple studies have demonstrated that long-read methods have better resolution than short-read methods for species and strain-level identification in microbiome analyses. For example, see here, here, and here. Overall, high-quality long sequences allow for better discrimination between closely related species in sequence-based microbial analyses.
Preprocessing
After sequencing, the raw reads are passed through a bioinformatics pipeline for preprocessing and analysis. Common pipelines include MetaGenome Rapid Annotation using Subsystem Technology (MG-RAST), Quantitative Insights into Microbial Ecology 2 (QIIME2), and Mothur.
Preprocessing raw sequencing data before full-scale analysis is critical for producing reliable and reproducible data. This typically involves performing an overall QC check of data integrity, filtering low-quality reads, and removing contaminants such as sequencing adapters and amplicon primers. Chimeric sequences (different sequences incorrectly joined during the PCR amplification step) also need to be removed because they could lead to errors in microorganism identification and diversity measurement. Demultiplexing is also necessary if it is not automatically handled by the sequencing platform, where barcode information is used to identify which sequences came from which samples so the sequences can be appropriately assigned back to the samples from which they were derived.
Operational taxonomic unit (OTU)/amplicon sequence variant (ASV) identification
Next, the sequences need to be converted into usable features for taxonomic classification. A major challenge is to distinguish real nucleotides from sequencing errors. It’s impossible to analyze each sequence individually, so OTU and ASV have been developed to simplify this process.
The OTU approach places sequences into groups, or “bins” based on similarity. Typically, a similarity threshold of 97% is used for optimal genus or species separation. This clustering can be reference-based (closed reference), reference-free (de novo), or a combination of both (open reference). While using reference sequence data is more computationally efficient, relying on databases of known sequences precludes identifying novel taxa. QIIME and Mothur are examples of pipelines that implement OTUs for 16S rRNA analysis.
OTU risks combining similar species into a single group, resulting in a loss of measured diversity. ASV is a newer method that addresses this issue by accounting for the frequency of each exact sequence. This is often referred to as “denoising” and relies on generating an error model for each run to identify sequences expected due to error or true biological variation. More precise and finer data resolution can be achieved because differences in a single nucleotide can be defined as a separate ASV. Tools that incorporate ASV include DADA2, DeBlur, and UNOISE3. And DADA2 has been verified for its good performance in dealing with the full-length 16S rRNA sequencing in the previous study.
Taxonomic assignment
The main goal of any microbiome sequencing experiment is to identify the microbiota composition in the samples. Reference databases include SILVA, Greengenes, and the Ribosomal Database Project, which are used with a classifier algorithm to find the best match for an OTU or ASV.
Downstream analyses: diversity, differential abundance, and functional prediction
Moving beyond classification, an actual measure of microbial diversity within and between samples also provides important insight into microbiome composition. Alpha diversity is the balance of microbes within a sample, including the total number of different microbes detected and their distribution. Beta diversity is the variation of microbiota between samples (e.g., treated versus control).
Many statistical tests can identify specific taxa that differ between samples. These range from simple t-tests to more advanced statistical models, such as those originally built for RNA-Seq data (DESeq2, edgeR), log-ratio-based methods (ANCOM, ALDEx2, DR/differential ranking), alternative mixture models based on zero-inflated Gaussian (Metagenome Seq), and balance-based (ratio) methods. With such a large number of methods available, it can be challenging to know which approach will provide the most reliable results. A recent study comparing the performance of 14 different methods recommends that researchers use multiple tools to get the most out of their data.
Simply knowing the microbial composition of a sample provides little information regarding the functionality of the microbial communities. Although this cannot be directly assessed with a single marker gene, a number of sophisticated tools (e.g., PICRUSt, Tax4Fun) infer the metagenome (the genomes of all microorganisms in a sample) and use this information to predict functional potential from databases of gene families and pathways.
Pros and cons of 16S amplicon sequencing
This sequencing is certainly the most cost-efficient method to capture microbiome diversity and is thus an excellent choice for projects with a large number of samples. Robust bioinformatics pipelines and reference databases are available, making analysis more straightforward. However, sequencing biases associated with PCR amplification and the assessment of only a subset of variable regions by the short-read method can lower taxonomic resolution. The higher resolution afforded by full-length 16S rRNA sequencing permits higher-level taxonomic classification but is costlier than the short-read 16S sequencing method. Additionally, 16S rRNA sequencing also only covers bacteria and archaea, so researchers who require a broader picture will need to consider this when designing experiments.
Shotgun metagenomic sequencing
Metagenomic sequencing offers an alternative to the marker-gene approach of 16S amplicon profiling. It covers the microbiome more comprehensively by sequencing all genetic material in a sample. This untargeted method is also known as “shotgun sequencing”. A major benefit of 16S sequencing is the ability to profile a broader spectrum of microbes, including viruses. It can also provide strain-level resolution, though this depends on the availability and quality of databases and reference genomes.
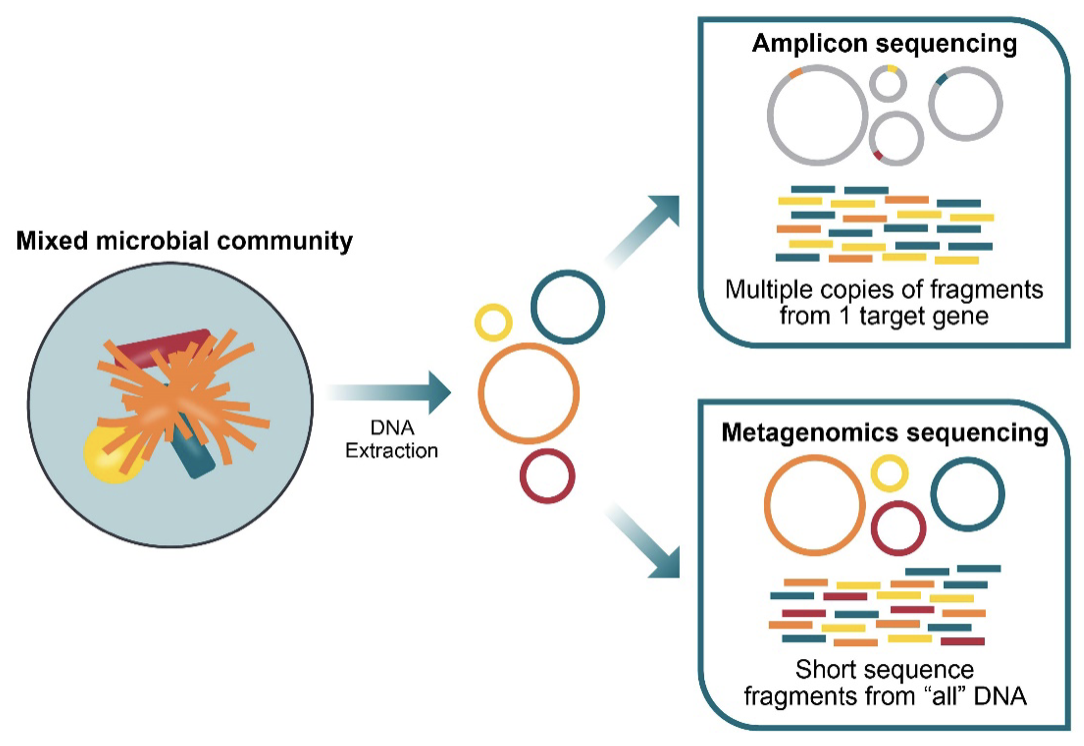
Figure 4. 16S amplicon sequencing vs metagenomics sequencing for microbiome analysis
Bioinformatic analysis of shotgun metagenomics data
The vast amounts of data generated by metagenomics sequencing makes bioinformatics analyses significantly more complex compared to 16S rRNA studies. Analyses of metagenomics data can be divided into two main categories: read-based and genome-resolved.
In the former, the raw data is preprocessed and mapped to reference genomes for taxonomic assignment. Classification may employ a binning approach that groups read-based on sequence composition or similarity to a genome database (e.g., the algorithms from Kraken2 and Centrifuge). While relatively straightforward, this is computationally intensive and depends heavily on the availability and quality of reference genomes. Alternatively, taxonomic assignment can rely on single or multiple marker genes (e.g., MetaPhlAn3). The reads are compared to a database containing informative gene sequences from specific taxa or phyla, which can be identified down to the strain level. Furthermore, the widely used marker gene-based pipeline HUMAnN has recently released its third iteration, which can accurately output presence/absence and estimates of the abundance of gene families and pathways to which microbial communities could potentially contribute.
Genome-resolved metagenomics analysis assembles reads into draft, or near complete, microbial genomes. These reconstructed metagenomes are used for analysis instead of a read-based analysis relying on reference genomes generated from sequencing isolated organisms.
A typical workflow for genome-resolved analysis involves the following steps:
- Data preprocessing (including a step to remove contaminating host reads)
- De novo assembly
- Genome binning to cluster sequence groups from the same organism
- Taxonomy, function, pathway profiling or other downstream analyses using the metagenome-assembled genomes (MAGs)
With the help of metagenome-assembled genomes (MAGs) yielded in genome-resolved metagenomic analysis, researchers have managed to identify the uncultivable microorganisms in humans and mice. Moreover, with all genes in MAGs annotated, researchers can identify the functions, pathways, and metabolisms of the microbiome more directly using the public protein domain, ontology, or metabolic pathway databases (e.g., GO, KEGG, CAZY, etc.). In addition, genome-resolved analysis also makes it possible to study the cis-elements in the genomes, like operons.
Pros and cons of metagenomic sequencing
The major benefit of metagenomic sequencing over 16S rRNA sequencing is that it obtains information regarding the entire genome. This allows for higher taxonomic resolution, more direct functional profiling, and the identification of novel/rare species. However, increased data comes at a cost. Shotgun sequencing is much more expensive and requires specialized bioinformatics expertise. It is a good choice for researchers looking to examine a smaller number of samples but in more depth than what is offered by amplicon sequencing.
Metatranscriptomic sequencing
Genomic sequencing constitutes the majority of published studies on the microbiome. However, DNA-level investigations are unable to determine which microbes are actively involved in biological processes. Metatranscriptomic sequencing presents a powerful opportunity to assess how the microbiome responds to environmental changes at the gene expression level. By employing RNA-Seq, researchers can bridge this gap and obtain comprehensive gene expression profiles that offer direct insights into the functional activities of living microbes.
Metatranscriptomic sequencing shares many similarities with shotgun metagenomics sequencing but requires additional considerations. Notably, the presence of abundant ribosomal RNA can significantly impact mRNA sequencing coverage. Therefore, these contaminants must be removed through ribodepletion during sample preparation or digitally eliminated during post-sequencing analysis.
The bioinformatic workflow for metatranscriptomics can be divided into read-based (e.g., MetaTrans) or assembly-based (e.g., SqueezeMeta) approaches. The selection of the workflow depends on the project goals and the availability of high-quality reference genomes. Since the primary objective of metatranscriptomic studies is to assess microbial activities, functional annotation is essential in the analysis. Several gene annotation tools compatible with transcript data are available, enabling the mapping of reads at the pathway level (e.g., FMAP, DIAMOND, MetaCLADE). Additionally, various pipelines and software have been developed to address the complexities of metatranscriptomic data. For instance, the HUMAnN3 pipeline accurately profiles the presence/absence, abundance, and activity of metabolic pathways in a microbial community. Similarly, MEGAN (MEtaGenome ANalyzer) simplifies the comparison, analysis, and exploration of metatranscriptomic data. Furthermore, tools like Rockhopper and Trinity assist in de novo transcriptome assembly, providing avenues for analysis when reference genomes are unavailable.
Although challenging due to transcripts originating from diverse organisms and the presence of shared genes among related but different organisms, RNA-level sequencing of the microbiome has successfully revealed microorganism-specific activities. Moreover, the future of metatranscriptomics appears promising, given the continuous technological advancements in tools and algorithms for analyzing metatranscriptomic data. As these new tools are deployed, they will accelerate our ability to efficiently identify the biologically active fraction of microbiomes.
Conclusion
Modern sequencing has revolutionized the field of microbiome research. These technologies offer in-depth taxonomic characterization, functional prediction or more direct assessment of activity within complex communities of microorganisms. Advances in bioinformatic analysis, long-read technology, and sequencing cost reduction will ensure new discoveries are possible and move the field forward.