The Transition from In Vitro to In Vivo Oncology Drug Development
At some point during your oncology drug development, you’ll want to move from in vitro studies to in vivo validation. A big choice to make at this point is which in vivo models you’re going to use. Historically, initial in vitro screening would be via panels of cancer cell lines, with transition to the in vivo models of interest as their cell line derived xenograft counterparts.
Now, more predictive patient-derived xenograft (PDX) models are often preferred for in vivo testing, so there’s the question of which are the right models for you and your agent. You could try an initial large scale screen using PDX models, but as in vivo models PDX aren’t really amenable to high-throughput screening (HTS) due to costs and time/labor for generation and maintenance.
The Development of PDX-Derived Organoids
An alternate method for PDX model selection is by using models derived from PDX for in vitro screening, such as PDX-derived organoids (PDXO).
Tumor organoids, including PDXO, are a revolutionary system within oncology drug development, with many applications across library screening and immunotherapy assessment. They are 3D in vitro culture systems developed from stem cells which recapitulate the complexities seen across a range of human cancer tissues. This includes cancer stem cells (CSCs) and their downstream differentiated progeny, as well as capturing all the key phenotypic and genetic features seen in parental tumors.
Tumor organoids are derived either directly from patient tumor tissue (patient-derived organoids or PDO), or as PDXO from PDX which expands PDO collections and their included genotypes/phenotypes.
The Benefits of PDXO including PDX Selection
There are multiple benefits of using PDXO models. Since large PDX panels already exist, PDXOs can be rapidly established and expanded for large-scale drug screens. This combines the benefits of well-characterized and predictive PDX models, with an in vitro platform amenable to HTS.
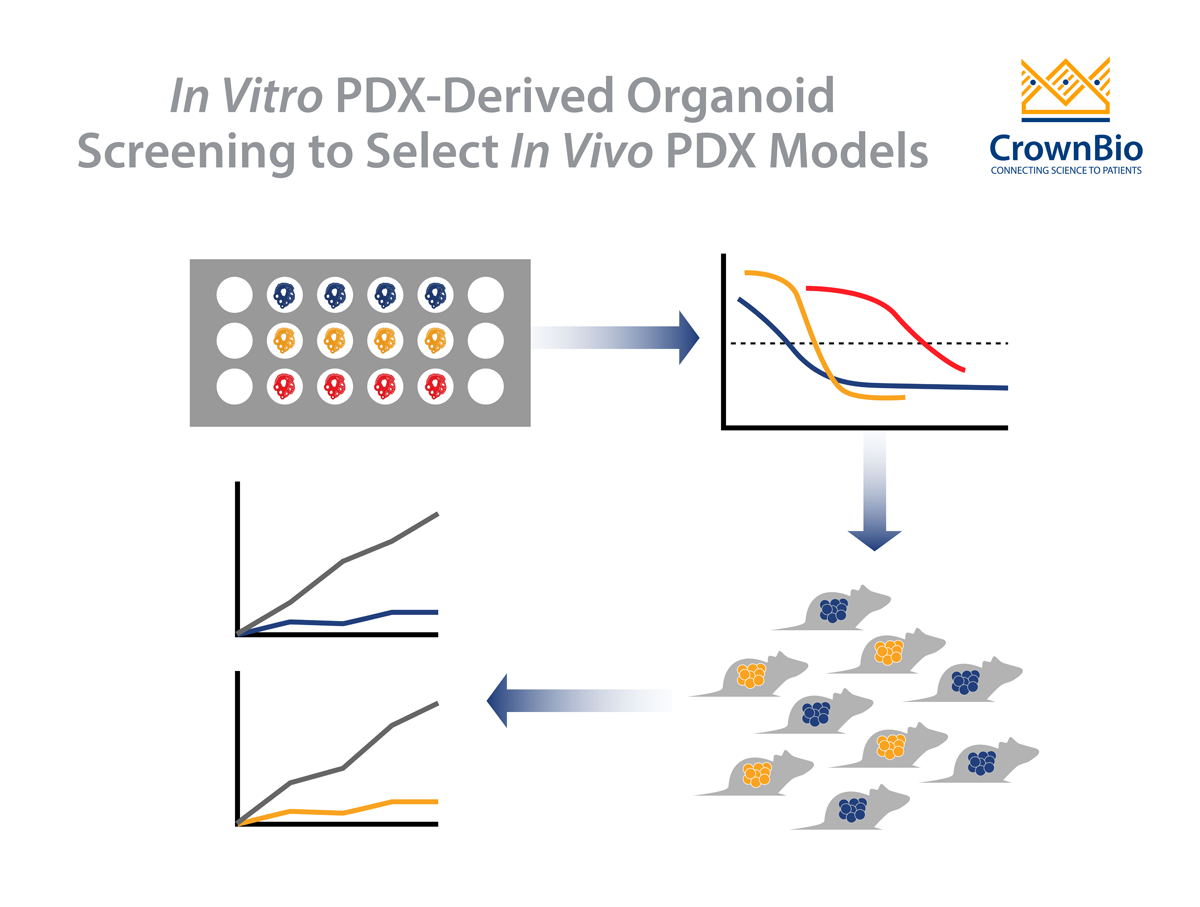
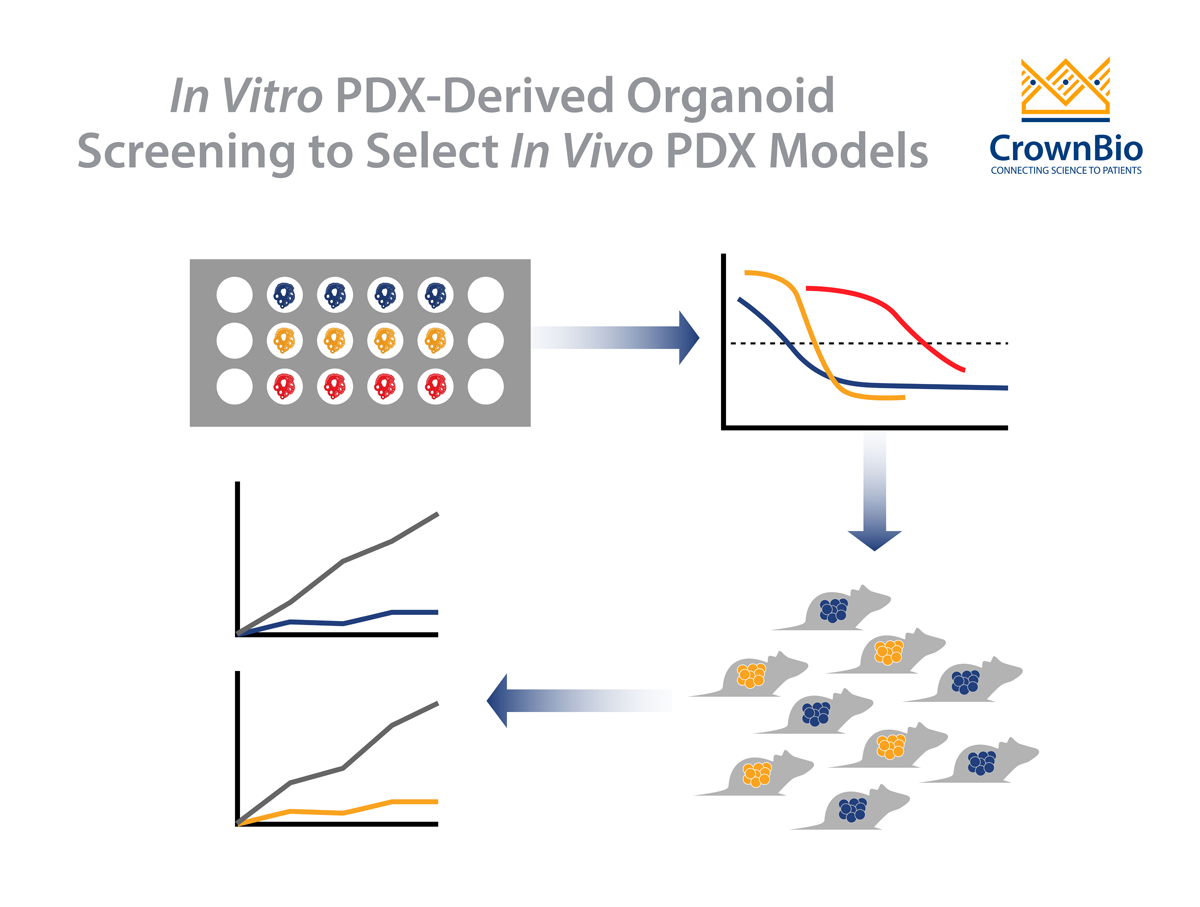
An added benefit is the availability of the parental PDX. This allows a simple workflow, with drug potency and efficacy tested in vitro on highly patient-relevant PDXO, with follow on in vivo validation studies:
Perform a PDXO in vitro screen evaluating a large number of models, effectively performing clinical trials in a dish
Use the data to identify subsets of corresponding responders/non-responders
Translate to the matched PDX model for validation of response in vivo
This ensures a much more informed and efficient transition from early stage in vitro efficacy testing to late stage in vivo validation studies to confirm efficacy and PK/PD effects. The transition is supported by data showing that PDXO in vitro drug response mirrors the corresponding PDX in vivo response.
The high level of characterization of the PDX models selected also enables further downstream applications in in silico analysis leading to the discovery of biomarkers of response.
Conclusion
Selecting the right in vivo PDX is possible by using PDX-derived organoids, and is one of the many new applications brought to oncology drug discovery from tumor organoid generation. It’s also become possible to reduce and optimize animal usage and accelerate the preclinical drug development workflow. By combining the benefits of in vitro tumor organoids with their in vivo PDX counterparts, a unique matched platform is provided, leading to more successful transition from early in vitro assessments to late-stage in vivo validation studies.
Cite this Article
Barbeau, J., (2020) How to Select the Right In Vivo PDX Using PDX-Derived Organoids (PDXO) - Crown Bioscience. https://blog.crownbio.com/select-the-right-in-vivo-pdx
Imagine standing on the brink of a breakthrough in cancer treatment, only to be halted by one daunting barrier: drug resistance. In this episode of Cr…
Oncology drug approvals in H1 2025 In the first half of 2025, the FDA’s Center for Drug Evaluation and Research (CDER) approved a total of 16 novel dr…
Often referred to as “magic bullets” or “biological missiles”, antibody-drug conjugates (ADCs) are one of the most promising advances in targeted canc…