 This week in Boston we’re presenting data on our new analysis platform and preclinical technologies. This post recaps all the new information on genomic and proteomic profiling of PDX models, our automated biomarker discovery platform, and deep proteomics in syngeneic models to uncover anti-PD-1 mechanism of action.
This week in Boston we’re presenting data on our new analysis platform and preclinical technologies. This post recaps all the new information on genomic and proteomic profiling of PDX models, our automated biomarker discovery platform, and deep proteomics in syngeneic models to uncover anti-PD-1 mechanism of action.
Comparative Analysis of Proteomic and Transcriptomic Profiles of a Panel of Gastric Carcinoma PDXs (AACR-NCI-EORTC 2019, Poster A025)
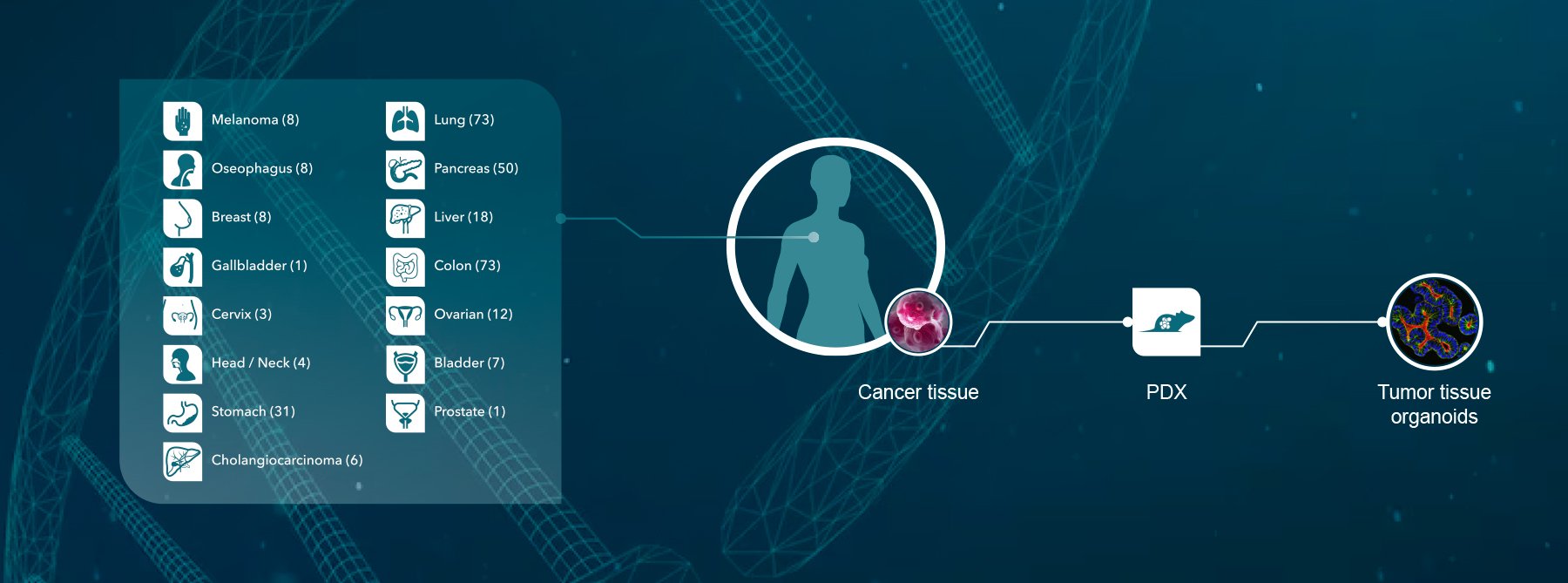
 Patient-derived xenografts (PDX) are a predictive preclinical model for translational oncology research. To maximize their full potential including for biomarker discovery, comprehensive genomic, proteomic, and pathological annotation of baseline and treated PDX models is needed.
Patient-derived xenografts (PDX) are a predictive preclinical model for translational oncology research. To maximize their full potential including for biomarker discovery, comprehensive genomic, proteomic, and pathological annotation of baseline and treated PDX models is needed.
Previously, we published on using gastric carcinoma PDX genomic profiles to identify biomarkers predictive of cetuximab response. Here, we analyzed the genomic and proteomic profiles of this gastric cancer PDX panel to guide the use of these annotations in translational research.
Correlating PDX Genomic and Proteomic Profiles
For this new study, 55 gastric cancer PDX were profiled by RNAseq and whole exome sequencing (WES), and 48 of the models were also subjected to whole proteomic analysis.
We found that there’s moderate correlation between mRNA expression and protein expression among the 48 PDX models. Further analysis revealed that genes with high mRNA expression and protein expression are enriched in protein translation and ribosome. This contrasts with genes with low mRNA expression and high protein expression, which are enriched in histones and protein folding/misfolding.
In conclusion, poster A025 shows how transcriptome and proteomics data is integrated to yield more informative and reliable biomarkers.
An Automated Biomarker Discovery Platform Based on In Vitro Pharmacology Raw Data (AACR-NCI-EORTC 2019, Poster A026)
 In vitro models such as cell lines and organoids are commonly used in cancer pharmacology studies to assess drug efficacy and pharmacodynamics. Genomic features of the models are also correlated with efficacy to help uncover potential predictive biomarkers to improve patient stratification. Biomarker discovery, however, is often complex and labor intensive and there’s only a few robust tools available to automate the process.
In vitro models such as cell lines and organoids are commonly used in cancer pharmacology studies to assess drug efficacy and pharmacodynamics. Genomic features of the models are also correlated with efficacy to help uncover potential predictive biomarkers to improve patient stratification. Biomarker discovery, however, is often complex and labor intensive and there’s only a few robust tools available to automate the process.
To help improve biomarker discovery, we’re developing an automated platform for in vitro preclinical pharmacology studies. A variety of machine learning algorithms and statistical methods will be combined with software for data manipulation, calculation, and graphical display. The platform will enhance preclinical study efficiency and provide valuable insights for precision medicine.
Developing A Novel User-Friendly Biomarker Discovery Platform
Poster A026 includes example datasets, showing high drug sensitivity prediction accuracy using signature genes and differentially activated pathways.
Overall, our novel automated biomarker discovery platform will enable raw data uploads, data conversions, efficacy overview, and biomarker discovery analysis as well as data report generation for in vitro datasets.
Investigating Mechanism of Action (MOA) of Anti-PD-1 Treatment by Systematic Depletion of Different Immune Cells in Syngeneic Models (AACR-NCI-EORTC 2019, Poster C102)
 While immune checkpoint inhibitors have revolutionized anticancer therapy, only around 20% of patients actually respond to treatment. It’s becoming increasingly important to understand the mechanism of resistance to such therapies to extend durable responses to more patients.
While immune checkpoint inhibitors have revolutionized anticancer therapy, only around 20% of patients actually respond to treatment. It’s becoming increasingly important to understand the mechanism of resistance to such therapies to extend durable responses to more patients.
In this study we treated syngeneic tumor bearing mice with anti-PD-1 in combination with various immune cell depletion to understand the efficacy contribution of different immune lineages. Tumor growth curves, tumor growth inhibition, and pharmacodynamics as well as effectiveness of immune depletion were evaluated. In addition, tumor samples were processed for proteomics analysis (in collaboration with Biognosys).
Understanding Anti-PD-1 Response Through Immune Cell Depletion and Deep Proteomics
CD8+ T cell depletion weakened anti-PD-1 efficacy in 3 of the 4 models tested, and CD4+ T cell depletion induced varying effects across models, suggesting different MOAs model by model. The flow cytometry readouts after treatment largely correlated with antitumor efficacy. Interestingly, deep proteomic analysis revealed part of the MOA that led to the difference in CD4 depletion between MC38 and Hepa 1-6 models.
In conclusion, Poster C102 shows how immune cell lineages act differently upon anti-PD-1 blockade dependent on specific tumor microenvironments, and how proteomic analysis combined with efficacy data can provide further insight into MOA.
Read More on AACR-NCI-EORTC 2019
For more on our AACR-NCI-EORTC19 posters, including organoids and advances in our in vivo models.