Many agents in cancer research target specific molecules or pathways, meaning they are only effective in a certain subset of patients. Finding another use for an already developed drug can cut down on early stage research and offer new hope to other patient groups. This could be happening soon in leukemia, where preclinical research has potentially uncovered a new use for PARP inhibitors.
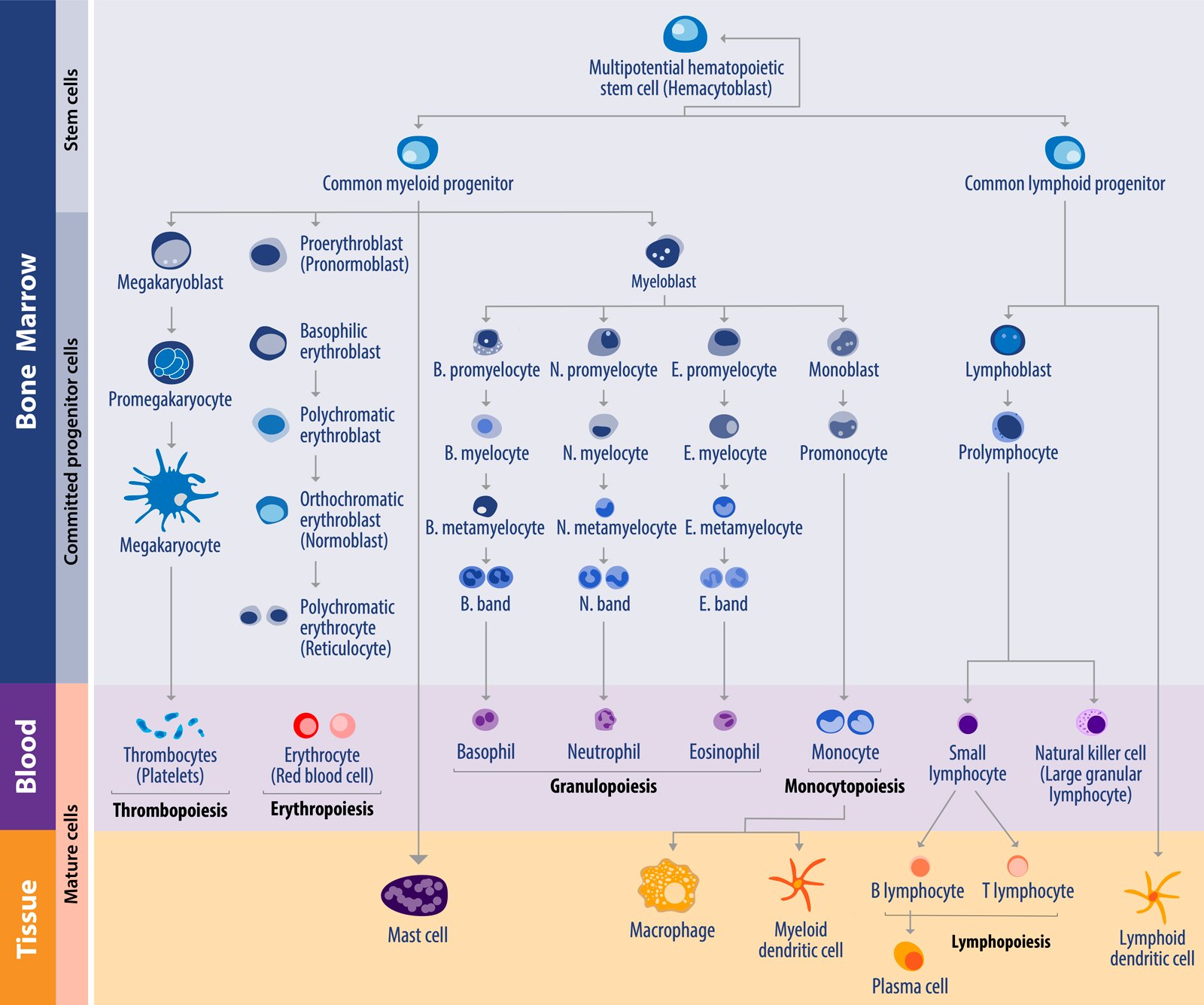
Leukemia is a group of cancers that start in early blood-forming cells and can be an acute or chronic disease. While chronic leukemia tends to be a disease of the elderly, acute leukemia is common in children and teenagers, and accounts for almost 1 out of 3 childhood and adolescent cancers. Around three quarters of these cases are acute lymphoblastic leukemia (ALL), with the remaining cases mainly being acute myeloid leukemia (AML). It is estimated that around 3,000 children a year in the US are diagnosed with leukemia, and the disease is the leading cause of death in children up to age 14.
Treatment of children with leukemia is intensive, usually starting with chemotherapy, and potentially also with a bone marrow transplant and radiotherapy. However, the ‘cure rate’ (defined as surviving for at least 5 years without cancer) from standard chemotherapy alone ranges from around 60 to 95% for children with ALL and only 40 to 50% for children with AML. New treatments are needed to improve these rates, and to try and decrease the harsh side effects that chemotherapy or transplantation bring to children’s lives.
A new treatment option for leukemia may have emerged in PARP inhibitors. These agents are mainly known for their action in breast and ovarian cancers, but are also in clinical trials for leukemia in adults and children. PARP inhibitors generate DNA strand breaks which cannot be repaired by cancer cells defective in homologous recombination, a DNA double strand break repair pathway. When PARP inhibitors are used in these deficient cell lines it leads to cell death, termed synthetic lethality. A paper published recently in Cell Reports has identified a new link between DNA repair and leukemia formation, which might hit the mark on how PARP inhibitors could be best used in leukemia. The study focused on the Runx family, which are transcription factors that regulate cell differentiation. The RUNX1 protein is one of the most frequently mutated in human leukemia, and RUNX3 is also associated with development of the disease.
The study showed that Runx1;Runx3 double knockout mice had a lethal phenotype due to bone marrow failure and a myeloproliferative disorder. These clinical findings are contradictory to each other, and researchers remarked that they resembled inherited bone marrow failure syndromes (such as Fanconi anemia), which develop due to defects in DNA repair proteins.
The researchers wondered if DNA repair deficiencies could also be helping to cause RUNX dependent leukemia. Further study of the double knockout mice did indeed find an impairment in DNA repair – a protein called FANCD2 (which is involved in Fanconi anemia) was not being recruited to DNA damage foci involved in homologous recombination, meaning that this DNA repair process could not be fully carried out. The research showed that RUNX proteins play a critical role in FANCD2 response and recruitment following DNA damage, and the loss and dysfunction of RUNX proteins leads to DNA repair defects.
This led the authors to treat cells with dysfunctional RUNX1 and RUNX3 with PARP inhibitors, to see if the effects seen in other homologous recombination defective cells could be repeated here. The cells were sensitive to treatment with PARP inhibitor, which was increased if the PARP inhibitor was combined with the chemotherapy agent mitomycin c. This shows that the synthetic lethal effects of PARP inhibition and MMC exposure could potentially be considered as a new treatment option for targeting RUNX-associated leukemias.
Crown Bioscience hopes that this repurposing of PARP inhibitors can help in the fight against leukemia in both adults and children, and looks forward to further research in this area. We support leukemia research through the use of our clinically relevant patient-derived xenograft models, HuKemia™, for AML and ALL as well as through our ex vivo models for AML, ALL, and chronic forms of the disease, available for oncology drug discovery and translational sciences. Crown Bioscience patient-derived xenograft AML models encompass different subtypes and phenotypes - our AM7577 model is of the M5 subtype, with IDH2 mutation (the only commercially available patient-derived xenograft model of this type), is FLT3-IDT+, and has a more differentiated phenotype. Our AM8096 patient-derived xenograft model is of the M2 subtype, which is less differentiated with leukemic cells mostly limited to the bone marrow.
Contact us today at busdev@crownbio.com to discover how we can transform your leukemia research through patient-derived xenograft model technology.