 We presented the latest data on our PDX models at this year’s AACR Annual Meeting in Chicago, with new studies on DLBCL models with diverse genotypes for BTK inhibitor testing, comparison of WES and RNAseq in PDX models, and on using PDX as a new model system to investigate the tumor microenvironment (TME).
We presented the latest data on our PDX models at this year’s AACR Annual Meeting in Chicago, with new studies on DLBCL models with diverse genotypes for BTK inhibitor testing, comparison of WES and RNAseq in PDX models, and on using PDX as a new model system to investigate the tumor microenvironment (TME).
AACR 2018 Poster 2167: Efficacy Assessment of the BTK Inhibitor Ibrutinib in De Novo and Viral-Induced B Cell Lymphoma
 Bruton's tyrosine kinase (BTK) is a major oncogenic driver for various B cell-derived lymphoid cancers. Ibrutinib, a BTK inhibitor, has been approved for a range of malignancies (e.g. mantle cell lymphoma, chronic lymphocytic leukemia) and as also shown encouraging efficacy in the ABC subtype of defuse large B cell lymphoma (DLBCL).
Bruton's tyrosine kinase (BTK) is a major oncogenic driver for various B cell-derived lymphoid cancers. Ibrutinib, a BTK inhibitor, has been approved for a range of malignancies (e.g. mantle cell lymphoma, chronic lymphocytic leukemia) and as also shown encouraging efficacy in the ABC subtype of defuse large B cell lymphoma (DLBCL).
To allow further testing of ibrutinib in DLBCL, preclinical models need to fully recapitulate the complex genetic background of the clinical disease. To meet this need, we’ve developed a panel of DLBCL PDX models which capture the diverse genotypes of de novo DLBCL.
- MYD88L265P/CD79BY197N double mutants
- MYD88L265P single mutant, plus wild type models)
- as well as EBV-transformed B lymphoma PDX models.
This poster details the validation and treatment of these models with ibrutinib, linking genetic characteristics to response.
All the background information for the models, as well as histopathology of the model panel, is shared. EBV-transformed B lymphoma PDX models share similar histopathology with de novo DLBCL, but have distinct molecular pathology signatures and different pathogenesis. Clustering of the EBV+ and EBV- models is shown, with gene set enrichment data.
Genetic Characteristics Play a Large Part in the Response to Ibrutinib
Genetics play a major role in determining a model’s response to ibrutinib. In the double mutant de novo DLBCL PDX models, moderate to robust efficacy is observed. This suggests that CD79B activating mutation is a predictive biomarker for chronic activation of BCR signaling, and of a generally good response to BTK inhibitors.
Interestingly, the MYD88 single mutant model (where the disease is likely driven by constitutive activation of MYD88 signaling) also showed response to ibrutinib when dosed in drinking water.
Nine wild type PDX were tested, with four models testing partially responsive or sensitive to Ibrutinib and five resistant to treatment. All five resistant models are EBV+, as verified by RNAseq.
Western Blot Analysis Following Ibrutinib
Finally, we shared western blot analyses for the models following ibrutinib treatment. No BTK phosphorylation and low t-BTK were found in EBV+ lymphoma, compared with the de novo models where ibrutinib highly inhibited the phosphorylation of BTK. It is likely that the pathogenesis of EBV-transformed models is associated with other immune deficiencies, such as interferon signaling, but not BCR.
To summarize this poster, our DLBCL and EBV-induced lymphoma PDX models provide a valuable preclinical platform for evaluating BTK inhibitors, as well as for future drug discovery efforts on other targets in the BCR and MYD88 pathways, such as PI3K, SYK, and IRAK4.
AACR 2018 Poster 1051: Mutation Detection in Patient-Derived Xenografts by Whole Exome Sequencing and Transcriptome Sequencing
 Various techniques are used to characterize PDX models, including:
Various techniques are used to characterize PDX models, including:
- RNAseq, for transcriptomic profiling to obtain data on gene expression, RNA-level mutations, and gene fusion.
- Whole Exome Sequencing (WES), to infer DNA-level mutations and copy number.
Poster 1051 compared mutation detection between RNAseq and WES across a selection of PDX models with both datasets available. Over 230 models met these criteria, covering twenty cancer types and large panels of colorectal, gastric, head and neck, and liver cancers.
The poster presents data comparing the two techniques, starting with the data sizes for each WES and RNAseq sample. Medians are around 15 GB and 12 GB for WES and RNAseq, respectively; therefore, if both are with the same sequencing depth, WES samples should contain more information than the RNAseq samples.
WES Provides a More Complete View of Mutations Due to the Fact that it Amplifies at the DNA Level
We verified how many variants each method detected at different quality levels (total variants, high-quality variants, or low-quality variants). Both technologies detected the vast majority of high-quality variants, with WES detecting more high-quality variants than RNAseq.
Around 48% of mutations detected by WES were also detected by RNAseq. For high-quality mutations, the fraction is approximately 43%. By comparison, 99% of the mutations detected by RNAseq based mutations are also captured by WES. This is due to WES amplifying exons at the DNA level and only being affected by experimental variations. Mutation ratios also showed a good correlation between WES and RNAseq.
In summary, both sequence analysis techniques have their benefits, but WES is able to give a more complete view of mutations due to the technique amplifying at the DNA level.
AACR 2018 Poster 1016: Transcriptomic Analysis of Bulk Tissues from a Large PDX Collection as a Novel Platform for Discovering New TME Targets/Drugs
 Poster 1016 brings our EORTC 2017 short talk presentation to a new audience, looking at how PDX can be used as surrogate models for studying the tumor microenvironment (TME).
Poster 1016 brings our EORTC 2017 short talk presentation to a new audience, looking at how PDX can be used as surrogate models for studying the tumor microenvironment (TME).
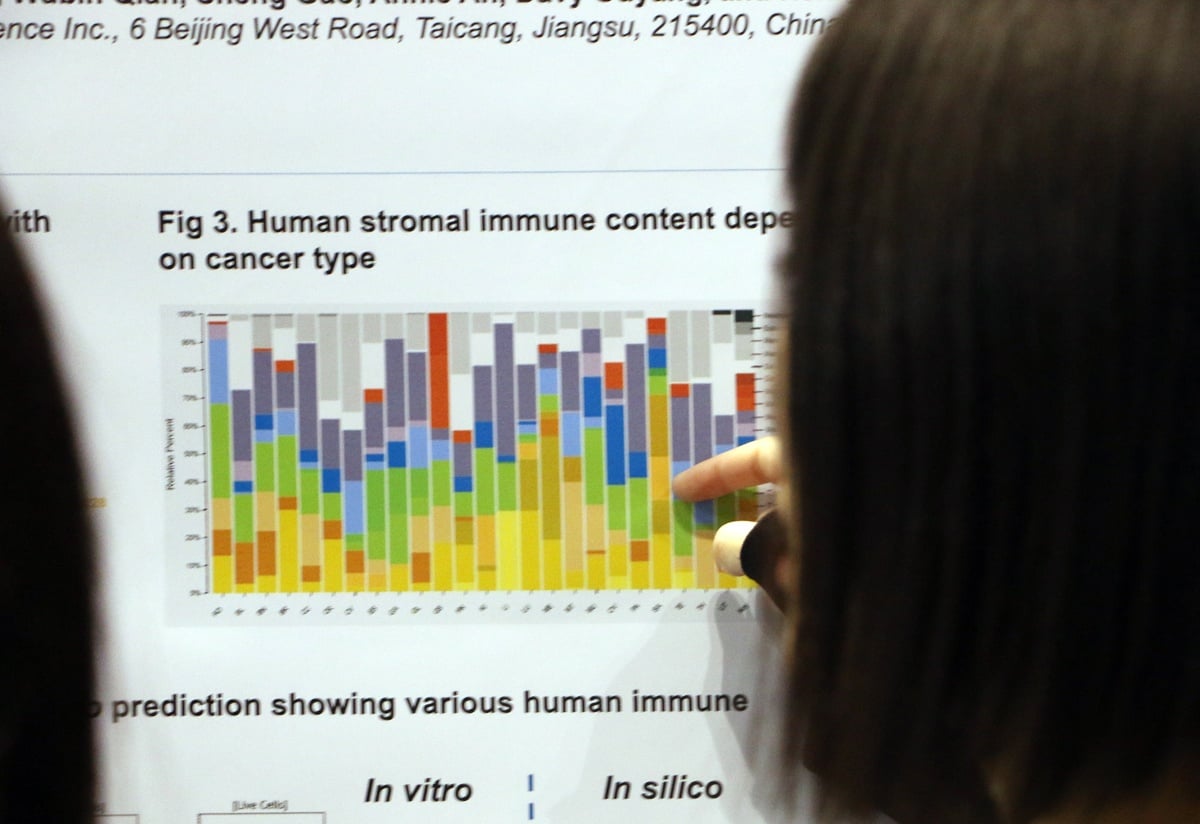
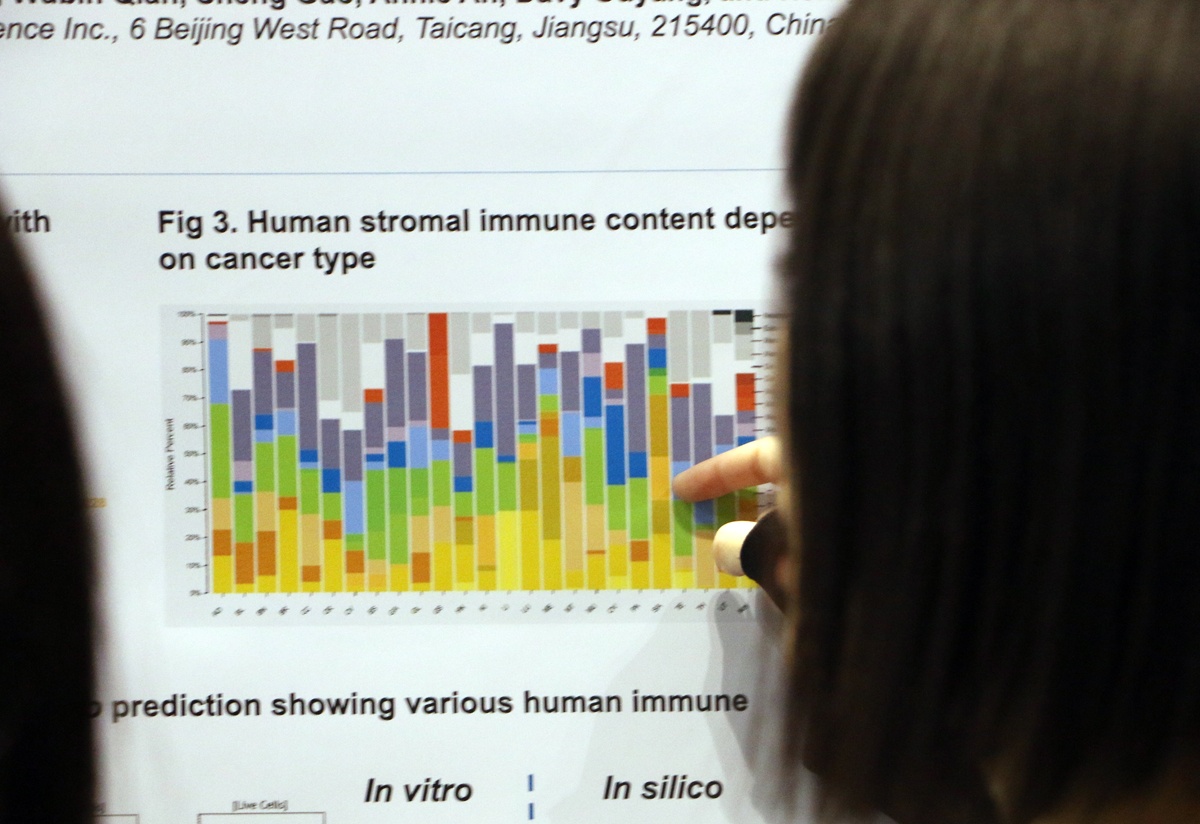
TME heterogeneity plays a major role in cancer progression and response to anticancer agents, particularly immunotherapies. However, studying the TME has proven to be especially challenging due to the difficulty in separating stroma and tumor cells, either physically via microdissection or in silico via bioinformatics. Here we look as using PDX to study the TME, as human and mouse content can be readily separated in these models.
The analysis looks at the RNAseq of around 1,600 tumor tissues from subcutaneous PDX models and the resulting analysis.
We shared the study schematic on our poster, explaining how whole transcriptome sequencing was performed, with reads aligned to human and mouse reference genomes to distinguish human and mouse content. A human-mouse gene correlation analysis was performed to construct cross-species expression networks. Overall, we found that the average mouse-to-human sequencing read ratio was approximately 11%, which is consistent with previous reports.
Deconvolution analysis of the human and mouse gene expression data showed that our analysis identified all types of adaptive and innate immune cells in mouse stroma and a variety of human immune components. The corresponding fractions were shown to vary across both cancer subtypes and individual models.
PDX Model Take Rate Increases when Interspecies Interactions are Lower
The independency of interspecies network sub-clusters, and its effects on PDX establishment and take rate, was also studied. Our co-regulation analysis identified a very high number of intra-species interactions and a much smaller number of inter-species interactions, which vary greatly among different cancer types.
Reverse-correlation between the number of interactions and take rate is shown across our PDX collections. Pancreatic cancer has the highest take-rate and lowest number of interactions, which interestingly also has the highest KRAS mutation rate (>90%). This suggests a role for KRAS mutation in tumor growth independency on TME, which is further confirmed for KRAS mutant CRC.
Overall, this poster shows that PDX models allow straightforward in silico separation of human and mouse content, supporting TME and tumor-stroma interaction studies.
Watch this space for more on AACR 2018, with featured posts on humanized mouse models, murine homograft models, ex vivo and in vitro assay platforms, and microbiome studies.